Levitra enthält Vardenafil, das eine kürzere Wirkdauer als Tadalafil hat, dafür aber schnell einsetzt. Männer, die diskret bestellen möchten, suchen häufig nach levitra kaufen ohne rezept. Dabei spielt die rechtliche Lage in der Schweiz eine wichtige Rolle.
Hkust institutional repository
International Journal of Neuropsychopharmacology (2011), 14, 1247–1256.
f CINP 2011
From understanding synaptic plasticity to thedevelopment of cognitive enhancers
Zelda H. Cheung and Nancy Y. Ip
Division of Life Science, State Key Laboratory of Molecular Neuroscience and Molecular Neuroscience Center,Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
Accumulating evidence reveals that synaptic dysfunction precedes neuronal loss in neurodegenerativediseases such as Alzheimer's disease. Intriguingly, synaptic abnormality is also implicated in a myriad ofpsychiatric disorders including depression. In particular, alterations in spine density and morphologyhave been associated with aberrant synaptic activity in these diseased brains. Understanding the mol-ecular mechanisms underlying the regulation of spine morphogenesis, synaptic function and plasticityunder physiological and pathological conditions will therefore provide critical insights for the develop-ment of potential therapeutic agents against these diseases. Here we summarize existing knowledge onsome of the molecular players in synaptic plasticity, and highlight how these findings from basic neuro-scientific research aid in the identification of novel drug leads for the development of therapeutics.
Received 26 August 2010 ; Reviewed 1 October 2010 ; Revised 5 November 2010 ; Accepted 19 November 2010 ;First published online 6 January 2011
Key words : Alzheimer's disease, BDNF, dendritic spines, depression, synaptic plasticity.
elucidating the pathophysiological mechanisms ofneurodegenerative diseases have focused on identify-
Since the first depiction of neurons by Ramon y Cajal
ing the signalling pathways that mediate the selective
more than a century ago, remarkable progress has
degeneration of the susceptible neuronal populations
been made to unveil the mystery behind the physi-
in these diseases. Interestingly, mounting evidence in-
ology, functioning and communication by these un-
dicates that loss of synapses precedes actual neuronal
ique cells. With the commencement of the molecular
death in Alzheimer's disease (AD) (Selkoe, 2002). In
and genetic era, knowledge on the molecular pathways
addition, advances in unravelling the molecular me-
implicated in the control of neuronal survival, synaptic
chanisms of depression point unexpectedly to deregu-
transmission and synaptic plasticity have exploded.
lation in synaptic plasticity (Krishnan & Nestler, 2008 ;
As much as these findings reveal how the neuronal
Pittenger & Duman, 2008). It therefore appears that
circuitry mediates daily physiological function of
dysfunction in synaptic transmission and plasticity
the nervous system, they also provide essential infor-
may present a common pathogenic mechanism across
mation for deciphering the molecular pathophysiology
a broad spectrum of neurological disorders. This reali-
of various neurological disorders. For example,
zation underscores the significance of in-depth in-
aberrant synaptic transmission has been implicated
vestigations into the molecular control of synapse
in psychiatric disorders such as schizophrenia and de-
function and plasticity, as these findings are likely to
pression, whereby the level of neurotransmitters is
generate critical new insights into the future develop-
abnormally elevated or reduced, affecting neuro-
ment of therapeutics that may be applicable to multiple
transmission. On the other hand, studies aimed at
disorders harbouring synaptic failures. In this brief re-view, we summarize the current understanding of themolecular mechanisms underlying the control of syn-
Address for Correspondence : Professor N. Y. Ip, Division of Life
aptic function and plasticity in AD and depression, as
Science, Hong Kong University of Science and Technology,
examples of neurodegenerative disease and psychi-
Clear Water Bay, Hong Kong, China.
atric disorder. How this knowledge may be utilized to
Tel. : 852-2358-7304 Fax : 852-2358-2765Email :
[email protected]
identify novel drug targets will also be discussed.
Z. H. Cheung and N. Y. Ip
Aberrant synaptic function and plasticity as a
harbouring various mutations in APP, which lead to
common pathophysiological mechanism in AD and
elevated Ab generation, exhibit impaired synaptic
transmission and LTP (Parsons et al. 2007 ; Selkoe,2002 ; Wasling et al. 2009). Interestingly, loss of post-
Aberrant synaptic function
synaptic marker PSD-95 around Ab plaques in an
AD is the leading cause of dementia worldwide and
APP transgenic mouse is paralleled by reduction in
accounts for more than 50 % of dementia cases. Post-
pre-synaptic boutons, suggesting that synapse loss
mortem brains of AD patients are characterized by the
involves both pre-synaptic and post-synaptic elements
presence of extracellular aggregates composed pre-
(Spires et al. 2005). Ab treatment in vitro and in vivo
dominantly of b-amyloid (also known as Ab) and
intracellular neurofibrillary tangles of hyperphos-
mediated signals and LTP (Hsieh et al. 2006 ; Selkoe,
phorylated tau. Ab generation has been postulated as
2002 ; Wasling et al. 2009), and reduces surface ex-
the main culprit in the aetiopathology of AD, as sup-
pression of NMDAR subunit NR1 by promoting its
ported by its abundant presence in senile plaques and
internalization (Kurup et al. 2010 ; Snyder et al. 2005).
the observation that essentially all missense mutations
This is accompanied by diminished NMDAR current
identified in familial cases of AD result in elevated
and the downstream activation of transcription factor
production of Ab (Hardy & Selkoe, 2002). Ab is gen-
CREB (Kurup et al. 2010 ; Snyder et al. 2005). In ad-
erated from sequential cleavage of a transmembrane
dition, AMPAR removal was found to underlie
protein called amyloid precursor protein (APP) by
Ab-stimulated synaptic depression and spine loss
b-secretase and c-secretase (Wasling et al. 2009). Since
(Hsieh et al. 2006). Taken together, these observations
AD is a neurodegenerative disease in nature, earlier
indicate that Ab can directly impair synaptic functions
efforts have been directed at understanding the mol-
and plasticity in AD brains by interfering with gluta-
ecular mechanisms by which Ab generation is regu-
matergic synapses.
lated, and how Ab deposits lead to death of the
Another neurological disorder characterized by ab-
neurons. Nonetheless, subsequent evidence indicates
errant synaptic function is depression. Also known as
that decrease in synapse density precedes actual loss
major depressive disorder, depression is a psychiatric
of neurons in AD brains. More importantly, synapse
disorder characterized by low mood, irritability, an-
loss, rather than neuron loss, serves as a more accu-
hedonia, difficulty in concentrating and cognitive im-
rate correlate with cognitive decline in AD patients
pairment. While the cause is essentially unknown,
(Selkoe, 2002). An increasing number of studies have
earlier studies aimed at elucidating the mechanisms of
therefore focused on elucidating the effect of Ab on
action of antidepressants suggest that reduced trans-
synaptic functions.
mission of two monoamine neurotransmitters in the
Several lines of evidence indicate that Ab can di-
brain, namely 5-HT (serotonin) and noradrenaline,
rectly impair synaptic transmission in AD. Glutamate
is the major excitatory neurotransmitter in the brain.
(Krishnan & Nestler, 2008 ; Pittenger & Duman, 2008).
There are two subtypes of ionotropic glutamate re-
Interestingly, accumulating evidence suggests that
ceptors, namely the AMPA receptors (AMPARs) and
impaired synaptic plasticity may also play a role in
the NMDA receptors (NMDARs). While AMPARs are
depression. Indeed, depressed patients exhibit diffi-
directly activated by ligand binding and mediate fast
culty in declarative memory (Zakzanis et al. 1998).
excitatory transmission at the synapse, NMDAR acti-
Stress, which is a known precipitating and aggravat-
vation requires concurrent ligand binding and de-
ing factor of depression, has been demonstrated to
polarization of the post-synaptic neuron. This unique
disrupt hippocampal LTP in experimental animals, in
property of NMDARs renders them particularly im-
addition to inducing atrophy of hippocampus, a situ-
portant in synaptic plasticity. NMDARs function as
ation also observed in depressed patients (Calabrese
‘coincidence detectors ', allowing the selective, long-
et al. 2009 ; Chen et al. 2010 ; Pittenger & Duman, 2008).
term strengthening of synaptic transmission following
Expression of brain-derived neurotrophic factor
high-frequency stimulation of the synapse in the
(BDNF), a neurotrophic factor that is critical for syn-
cellular paradigm of long-term potentiation (LTP).
aptic plasticity, was reduced in the hippocampus of
Indeed, activation of NMDARs was demonstrated to
post-mortem brains of depressed patients (Karege
be essential for the generation of synaptic plasticity in
et al. 2005). In addition, antidepressant treatment
the brain (Lau & Zukin, 2007). Interestingly, Ab has
enhances the expression of BDNF and transcription
been observed to selectively affect glutamatergic
factor CREB, which have been demonstrated as
synapses (Wasling et al. 2009). Transgenic mice
pivotal players in long-term synaptic plasticity
Developing cognitive enhancers from basic research
(Chen et al. 2001 ; Thome et al. 2000). These observa-
that post-mortem brains of human patients with
tions collectively suggest that alterations in synaptic
severe stress and longitudinal depression exhibit
transmission and plasticity may contribute to the
reduced dendritic spine densities (Soetanto et al. 2010).
pathophysiology of depression.
In agreement with these findings, treatment withantidepressants fluoxetine or imipramine enhancespine density in rats (Ampuero et al. 2010 ; Chen et al.
Spine morphology anomaly
2008a ; Hajszan et al. 2005), implicating structural
The majority of the excitatory synapses are located on
changes in dendritic spines and synaptic abnormality
tiny protrusions on dendrites known as spines. Spines
in depressed patients.
are highly dynamic structures, with their size, shapeand density along dendrites under constant and attimes, rapid, regulation. While the more elongated
Molecular players in the regulation of synaptic
protrusions known as filopodia are regarded as im-
functions and spine morphogenesis
mature spines and are highly plastic ; stubby, mush-
A plethora of signalling pathways has been implicated
room-shaped spines are generally mature spines that
in the control of synaptic functions. Nonetheless, re-
are more stable in nature (Tada & Sheng, 2006).
cent studies have highlighted several molecular play-
Interestingly, spine morphogenesis is regulated by
ers that show promise in allowing identification of
synaptic activity, and has been postulated as the
novel targets for developing drugs that can reverse
structural basis of synaptic plasticity. LTP, for ex-
ample, is associated with an enlargement of spinehead and insertion of AMPARs at synaptic sites ; while
Glutamate receptors
long-term depression (LTD) involves shrinkage ofspines (Dillon & Goda, 2005 ; Lau & Zukin, 2007 ; Tada
Being the receptors for the major excitatory synapses
& Sheng, 2006 ; Zhou et al. 2004). Changes in spine
in the brain, it is no surprise that glutamate receptors
morphology require the coordinated regulation of ac-
are pivotal for the control of synaptic strength. The
tin cytoskeleton and also gene transcription (Ethell &
type and number of glutamate receptors present at a
Pasquale, 2005). Given the essential role of spine
synapse are under tight dynamic regulation and serve
morphogenesis in synaptic plasticity, understanding
as one of the most direct determining factors of syn-
how spine morphology is altered in various neuro-
aptic strength. Rapid changes in the number of gluta-
logical diseases will provide essential background
mate receptors are mediated by insertion or removal
knowledge on how synaptic dysfunction may be re-
of surface receptors at the post-synaptic densities.
Lateral movement of both AMPA and NMDA re-
In addition to directly impairing synaptic trans-
ceptors also contributes to the rapid changes in gluta-
mission and plasticity by interfering with glutamater-
matergic transmission at the synapse (Hanley, 2008 ;
gic synapses, Ab has also been demonstrated to affect
Lau & Zukin, 2007). Furthermore, AMPA and NMDA
spine morphogenesis. Ab treatment has been observed
receptors play distinct roles in the induction of LTP.
to reduce dendritic spine density in cultured neurons
NMDAR activation during the concurrent presence of
and also in transgenic animal models of AD (Knobloch
synaptic glutamate and post-synaptic depolarization
& Mansuy, 2008 ; Lacor et al. 2007 ; Shankar et al. 2007 ;
results in calcium influx through the NMDAR and
Spires et al. 2005). In particular, a recent study dem-
activation of secondary signalling messengers such as
onstrated that Ab produced from axons or dendrites
Ca
2+/calmodulin-dependent kinase 2 (CaMKII). The
lowers spine number and plasticity on nearby den-
subsequent AMPAR insertion mediates the increase in
drites (Wei et al. 2010). These observations suggest that
synaptic strength during LTP induction (Lau & Zukin,
loss of dendritic spines may also contribute to the
2007 ; Monti & Contestabile, 2009). The signalling cas-
cognitive deficits in AD patients.
cade initiated downstream of NMDAR activation also
Changes in spine morphology in depression are not
triggers gene transcription, which has been demon-
as well documented but evidence in support of this
strated to be critical for the late phase of LTP (Kelleher
possibility is beginning to emerge. Chronic stress was
et al. 2004). Aside from being indispensible for gluta-
observed to reduce dendritic complexity (Bloss et al.
matergic transmission and synaptic plasticity, recent
2010 ; Hains et al. 2009 ; Liston et al. 2006), in addition to
evidence suggests that AMPARs may also directly
decreasing spine density in rats (Chen et al. 2008b,
regulate spine morphogenesis. The extracellular
2010 ; Hains et al. 2009 ; Radley et al. 2006 ; Silva-Gomez
N-terminal domain of the AMPAR subunit GluR2 was
et al. 2003). In addition, a recent study demonstrated
found to enhance spine growth (Passafaro et al. 2003 ;
Z. H. Cheung and N. Y. Ip
Saglietti et al. 2007). Collectively these observations
early phase of LTP (Waterhouse & Xu, 2009). TrkB
underscore the essential role of glutamate receptors in
activation at the synapse also enhances local protein
the control of synaptic function and plasticity.
synthesis, an action that is required for the late phase
Interestingly, as much as glutamate transmission is
of LTP (Bramham, 2008). Indeed, BDNF stimulation
required for normal function of the neural circuitry,
and depolarization increases trafficking of TrkB
excessive activation of glutamate receptors during
and BDNF mRNA to the dendrite (Righi et al.
pathological condition can also impair synaptic func-
2000 ; Tongiorgi et al. 1997), further supporting an
tion and lead to excitotoxic neuronal loss. Energy fail-
involvement of local protein synthesis in synaptic
ure or impairment of glutamate transporters in AD
plasticity. On the other hand, proBDNF was found
brains may contribute to an abnormally high level
to facilitate LTD induction (Woo et al. 2005). Interest-
of extracellular glutamate, leading to excitotoxicity
ingly, cleavage of proBDNF to mature BDNF is el-
(Lipton, 2006 ; Parsons et al. 2007). In support of a role
evated by high-frequency stimulation, one of the
of excitotoxicity in AD, memantine, an uncompetitive
stimulation paradigms used to induce LTP in hippo-
NMDAR antagonist approved by the FDA for the
campal slice preparation (Nagappan et al. 2009). These
treatment of AD, was found to reduce neuronal loss
observations indicate that while BDNF signalling is
and also LTP impairment induced by Ab treatment or
crucial for synaptic function and plasticity, its role is
in animal models of AD (Lipton, 2006 ; Parsons et al.
complex and can be controlled at multiple levels.
2007 ; Rammes et al. 2008). The mechanism by which
Aside from being critical for LTP induction, BDNF
an NMDAR antagonist may reverse LTP impairment
has also been demonstrated to directly regulate spine
has not been completely elucidated, but it appears to
morphogenesis and has also been demonstrated to
involve reduction of tonic activation of glutamatergic
increase spine density (Amaral & Pozzo-Miller, 2007 ;
synapses (Lipton, 2006 ; Parsons et al. 2007 ; Rammes
Ji et al. 2005). Induction of spine head enlargement
et al. 2008). These findings reveal that although
by pairing of post-synaptic spike with glutamate un-
NMDARs may be a very attractive target for devel-
caging was found to require BDNF and local protein
opment of therapeutics, identifying compounds that
synthesis (Tanaka et al. 2008). Furthermore, when
inhibit pathological but not physiological glutamater-
dendritic targeting of BDNF mRNA is abolished by
gic transmission will be essential for ensuring the
expression of a truncated form of the long 3k-UTR
feasibility and applicability of these potential drugs.
BDNF mRNA, impairment in spine head enlargementand spine pruning are observed (An et al. 2008). Thesefindings demonstrated that BDNF also plays a role in
BDNF/TrkB signalling
spine morphogenesis, in part through regulation of
BDNF is a member of the neurotrophin family that
local protein synthesis (Fig. 1).
has been increasingly implicated in the regulation of
Extensive studies have demonstrated suppression
synaptic function and plasticity. Action of BDNF is
of BDNF signalling in AD and depression. Both ex-
mediated predominantly by receptor tyrosine kinase
pression of BDNF and TrkB are reduced in AD brains
TrkB, although it also binds with low affinity to p75.
(Schindowski et al. 2008). Interestingly, it was recently
BDNF is synthesized as proBDNF, which has long
demonstrated that BDNF deprivation enhances Ab
been regarded simply as the unprocessed form of
generation in hippocampal neurons (Matrone et al.
mature BDNF. Nonetheless, recent evidence reveals
2008). In agreement with this observation, BDNF was
that proBDNF is also secreted, and exhibits high-
found to reduce Ab production through activation of
affinity binding to p75 (Lu et al. 2005). While the
Sorting protein-related receptor with A-type repeats
neurotrophins were initially identified based on their
(SORLA), whose expression is reduced in sporadic AD
ability to maintain neuronal survival, BDNF was later
(Rohe et al. 2009). These observations reveal that BDNF
identified as being particularly critical for synaptic
signalling may be important for reducing Ab gener-
plasticity. Mice lacking BDNF exhibit impaired LTP
ation in AD brains. On the other hand, while an earlier
induction (Korte et al. 1995 ; Patterson et al. 1996).
observation of reduced BDNF level in the hippocam-
In addition, BDNF was found to be secreted at the
pus of depressed patients has triggered considerable
synapse in an activity-dependent manner. This local
interest in the hypothesis that BDNF deficiency may
elevation in BDNF results in TrkB activation at the
underlie the pathophysiology of depression, recent
synapses, and the subsequent initiation of down-
data are more controversial. For example, reduction of
stream signalling cascade modulates synaptic proteins
BDNF in the hippocampus is accompanied by elev-
to regulate the efficiency of synaptic transmission.
ated BDNF level in the nucleus accumbens (Krishnan
These modulations were found to be critical for the
& Nestler, 2008). In addition, while a reduction in
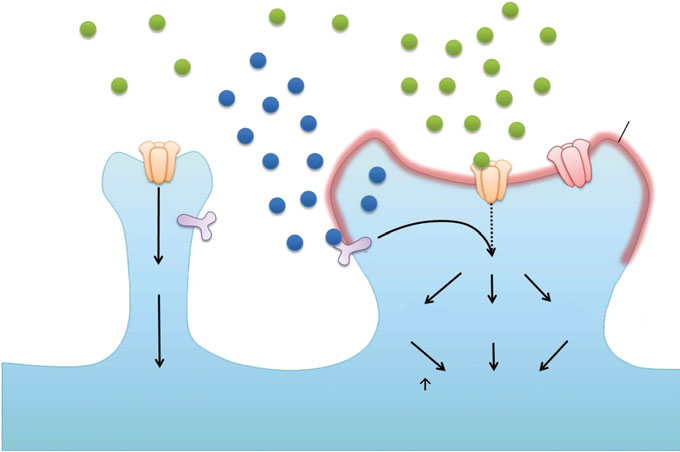
Developing cognitive enhancers from basic research
Local protein CREB-dependent
Spine size and number
No post-synaptic electrical activity
Post-synaptic electrical activity
Fig. 1. BDNF/TrkB signalling as a key regulator of synaptic plasticity. Synaptic activity increases the local release of BDNFat the synapse, which leads to dendritic targeting of BDNF and TrkB mRNA. Activation of TrkB then enhances expression ofsynaptic proteins through CREB-dependent transcription and local protein translation, resulting in enhanced membraneinsertion of AMPARs at synapses. The local protein synthesis by BDNF also promotes actin polymerization, contributing tostructural changes of dendritic spines and LTP formation. On the contrary, in the absence of post-synaptic activity, localBDNF/TrkB signalling and spine growth are both limited.
BDNF level fails to induce depression, BDNF is re-
of NR2B by calpain. This is accompanied by improved
quired for the efficacy of antidepressants (Calabrese
spatial learning in Cdk5 conditional knockout mice
et al. 2009 ; Krishnan & Nestler, 2008). Taken together
(Hawasli et al. 2007). These observations implicate
these observations reveal that while BDNF is probably
Cdk5 in the regulation of glutamate transmission and
involved in the pathophysiology of depression, its ac-
synaptic plasticity. Furthermore, Cdk5 has also been
tion is likely region-specific and additional studies will
demonstrated to regulate spine morphogenesis. Cdk5
be required to delineate its precise involvement.
was found to be required for ephrinA1-induced spineretraction through regulation of the downstream acti-vation of RhoA (Fu et al. 2007). In addition, Cdk5
Cyclin-dependent kinase 5 (Cdk5)
phosphorylates WAVE1 to inhibit actin polymeriz-
Cdk5 is a predominantly neural-specific serine/thre-
ation. This is associated with reduction of stubby-
onine kinase that has recently been implicated in the
shaped spines (Kim et al. 2006). These observations
regulation of synaptic function and plasticity (Cheung
reveal that Cdk5 is pivotal for the regulation of spine
et al. 2006 ; Lai & Ip, 2009). Cdk5 is activated upon
binding to its activator p35 or p39. Earlier studies
Cdk5 has long been implicated in the pathophy-
demonstrated that Cdk5 and its activators are ex-
siology of AD. In particular, cleavage of p35 into a p25
pressed at the synapse (Cheung et al. 2006). Evidence
fragment, which results in prolonged activation of
in support of a role of Cdk5 in the regulation of syn-
Cdk5, is associated with neuronal loss in various
aptic transmission came from the observation that
models of neurodegenerative diseases (Cheung et al.
Cdk5 was found to directly phosphorylate NMDAR
2006). Indeed, Cdk5 was initially identified as a tau
subunit NR2A, and inhibition of this phosphorylation
kinase (Kobayashi et al. 1993). In addition, neuronal
reduces NMDA-evoked current (Li et al. 2001). In ad-
loss and impairment in spatial learning are evident
dition, a recent study revealed that inhibition of Cdk5
in a mouse model with prolonged expression of p25
activity reduces activity-dependent internalization of
(Fischer et al. 2005). Interestingly, expression of
NMDARs by modulating phosphorylation of the
b-secretase BACE1 is also elevated in this strain of
NMDAR subunit NR2B by Src (Zhang et al. 2008).
mouse, leading to elevated production of Ab (Wen
Interestingly, Cdk5 was found to mediate degradation
et al. 2008). These studies collectively indicate that
Z. H. Cheung and N. Y. Ip
aberrant activation of Cdk5 may contribute to thepathophysiology of AD.
Explore disease mechanisms
Impaired BDNF/TrkB signalling
A role of Cdk5 in depression is not as solidly dem-
Cdk5 activity deregulation
onstrated. Nonetheless, Cdk5 has been observed to
Aberrant NMDA receptors activation
regulate dopamine signalling, the dysfunction ofwhich has been associated with the pathophysiologyof depression (Rakofsky et al. 2009). Cdk5 was dem-
Identify disease-related proteins – molecular targets
onstrated to phosphorylate DARPP-32, an importantplayer in dopamine signalling. Cdk5-mediated phos-
phorylation of DARPP-32 reduces dopamine-inducedactivation of PKA, thereby attenuating dopaminergicsignalling (Benavides & Bibb, 2004). These observa-tions suggest that Cdk5 may, through its modulation
Discovery of Drug Leads
of dopaminergic signalling, contribute to monoamine
imbalance in depressed patients. Further studies
will be required to address the precise role of Cdk5 indepression.
Identification of novel drug leads that target
Fig. 2. From basic research to drug discovery. Basic research
regulators of synaptic plasticity
focused on understanding the pathophysiologicalmechanism of neurological diseases will enable identification
Basic research focused on understanding the patho-
of molecular players that may serve as drug targets.
physiology of various diseases is crucial for the de-
Subsequent screening of compounds that selectively target
velopment of therapeutic interventions. Explicating
these molecules of interest will provide important basis for
the signalling pathways that are affected in the dis-
the development of novel therapeutics.
order will enable the identification of proteins thatmay be targeted to ameliorate symptoms or delay
trials have been performed with various NMDAR an-
disease progression. Through the uncovering of novel
tagonists but since physiological level of glutamate
molecular players, drug leads that directly target
transmission is pivotal for the normal function of the
these molecules can then be developed and tested
brain, many of the antagonists tested resulted in major
as potential therapeutics against the disease (Fig. 2).
side-effects and the studies were discontinued.
This approach has served as one of the most important
Memantine, currently the only NMDAR antagonist
strategies for effective design and development
approved for the treatment of AD, is unique as a non-
of drugs and treatment against various disorders.
competitive, relatively low-affinity, open-channel an-
Indeed, advances in understanding the molecular
tagonist that exhibits strong voltage-dependence and
mechanisms implicated in the pathophysiology of
fast off-rate (Lipton, 2006 ; Monti & Contestabile, 2009 ;
neurological disorders such as AD and depression
Rammes et al. 2008). This special property of meman-
have led to the development of the existing treatments
tine allows it to preferentially bind to NMDARs that
for these diseases. For example, the observed re-
are open for a prolonged period, without interfering
duction in cholinergic transmission in post-mortem
with the normal physiological function of the receptor
AD brains prompted the development of AChE
(Lipton, 2006 ; Monti & Contestabile, 2009 ; Rammes
inhibitors as therapeutic agents for AD (Monti &
et al. 2008). These features of memantine explain its
Contestabile, 2009). However, recent evidence has re-
clinical tolerance and suitability for treatment of ex-
vealed that inhibition of nicotinic acetylcholine re-
ceptors reduces Ab production and Ab-induced spine
The emerging involvement of aberrant synaptic
loss (Wei et al. 2010), raising the possibility that acti-
function and plasticity in AD and depression provided
vation of nicotinic acetylcholine receptors by increas-
important insights on identifying new molecular
ing acetylcholine level via AChE inhibition may be
players as potential drug targets. In particular, modu-
detrimental. On the other hand, the induction of ex-
lators of NMDARs that facilitate synaptic trans-
citotoxic death following Ab treatment and evidence
mission or LTP induction could potentially function
of excitotoxicity in AD brains led to the exploration of
as cognitive enhancers to limit cognitive decline in
NMDAR antagonists as potential neuroprotective
AD and depression. Interestingly, studies aimed at
agents against neuronal loss (Lipton, 2006). Many
elucidating the mechanisms of action of memantine
Developing cognitive enhancers from basic research
reveal that excessive NMDAR activation may also
pathophysiological machinery of various neurological
impair synaptic plasticity (Parsons et al. 2007), sug-
disorders, it is interesting to note that studies aimed at
gesting that other NMDAR antagonists may be devel-
explicating the mechanism of action of existing drugs
oped as cognitive enhancers to combat decline in
have also provided remarkable insights. For example,
cognitive functions. In light of the successful devel-
recent studies have revealed that neurogenesis in-
opment of memantine as AD therapeutics, it is im-
duced by antidepressants is critical for their thera-
portant to select for NMDAR antagonists that preserve
peutic effect (Krishnan & Nestler, 2008 ; Pittenger &
physiological function of NMDARs. Similarly, devel-
Duman, 2008). Interestingly, memantine has also been
opment of positive modulators targeting AMPAR
recently observed to induce adult neurogenesis in
trafficking and function may also prove to be
the hippocampus (Maekawa et al. 2009). In light of the
beneficial to ameliorating synaptic deficit in these
emerging involvement of neurogenesis in learning
disorders. On the other hand, the critical involvement
and memory (Deng et al. 2010), it will be important to
of BDNF in synaptic plasticity indicates that TrkB
explore if other modulators of neurogenesis may also
agonists may also help to reverse synaptic deficits in
be developed as cognitive enhancers.
AD and certain brain regions in depressed patients.
Direct infusion of trophic factors such as NGF as
therapeutics for neurodegenerative diseases have longattracted interest based on their neuroprotective effect.
We thank Ka-Chun Lok for his excellent help in
Nonetheless, inefficient crossing of the blood–brain
preparing the figures and Dr Amy Fu for critical
barrier and the non-specific effect of these trophic
reading of the manuscript. The study of N. Y. Ip and
factors have limited their applicability as therapeutics.
Z. H. Cheung was supported in part by the Research
It is therefore important to identify TrkB agonists that
Grants Council of Hong Kong (HKUST 6431/06M,
can cross the blood–brain barrier, in addition to de-
661109, 661309 and 1/06C) and the Area of Excellence
veloping methods that would allow precise delivery of
Scheme of the University Grants Committee (AoE/
the agonists. Interestingly, a recent study reported the
B-15/01) and the Hong Kong Jockey Club. N. Y. Ip and
identification of a small molecule BDNF mimetic that
Z. H. Cheung were Croucher Foundation Senior
reduces neurotoxin-induced neuronal loss in vitro, in
Research Fellow and Croucher Foundation Fellow,
addition to restoring motor learning following trau-
matic brain injury (Massa et al. 2010). This observationfurther supports the potential therapeutic efficacy of
Statement of Interest
TrkB agonists in ameliorating synaptic deficits inneurodegenerative diseases. Cdk5 inhibitors present
yet another potential drug target for the developmentof cognitive enhancer in light of its involvement in
AD pathology. In particular, since Cdk5 conditionalknockout mice exhibit enhanced spatial learning
Amaral MD, Pozzo-Miller L (2007). TRPC3 channels are
necessary for brain-derived neurotrophic factor to
(Hawasli et al. 2007), the development of Cdk5 in-
activate a nonselective cationic current and to induce
hibitors as therapeutics for AD will not only alleviate
dendritic spine formation. Journal of Neuroscience 27,
neuronal loss, but will also enable amelioration of
disease progression by targeting synaptic dysfunction
Ampuero E, Rubio FJ, Falcon R, Sandoval M, et al. (2010).
early on (Fig. 2). Collectively, these studies have
Chronic fluoxetine treatment induces structural plasticity
identified several new molecular players that may be
and selective changes in glutamate receptor subunits in
targeted to reverse synaptic and cognitive deficits in
the rat cerebral cortex. Neuroscience 169, 98–108.
AD, and other disorders where synaptic impairment
An JJ, Gharami K, Liao GY, Woo NH, et al. (2008). Distinct
may be implicated. Interestingly, there has also been
role of long 3k UTR BDNF mRNA in spine morphology
an increasing demand for cognitive enhancers as a
and synaptic plasticity in hippocampal neurons. Cell 134,
lifestyle drug for healthy individuals. While the ethical
Benavides DR, Bibb JA (2004). Role of Cdk5 in drug abuse
issues related to the consumption of cognitive en-
and plasticity. Annals of the New York Academy of Sciences
hancers remain a debated topic, it is plausible that
1025, 335–344.
these cognitive enhancers may also benefit healthy
Bloss EB, Janssen WG, McEwen BS, Morrison JH (2010).
Interactive effects of stress and aging on structural
Finally, while novel drug targets are identified
plasticity in the prefrontal cortex. Journal of Neuroscience
from basic research focused on elucidating the
30, 6726–6731.
Z. H. Cheung and N. Y. Ip
Bramham CR (2008). Local protein synthesis, actin dynamics,
Hardy J, Selkoe DJ (2002). The amyloid hypothesis of
and LTP consolidation. Current Opinion in Neurobiology
Alzheimer's disease : progress and problems on the road
18, 524–531.
to therapeutics. Science 297, 353–356.
Calabrese F, Molteni R, Racagni G, Riva MA (2009).
Hawasli AH, Benavides DR, Nguyen C, Kansy JW, et al.
Neuronal plasticity : a link between stress and mood
(2007). Cyclin-dependent kinase 5 governs learning and
disorders. Psychoneuroendocrinology 34 (Suppl. 1),
synaptic plasticity via control of NMDAR degradation.
Nature Neuroscience 10, 880–886.
Chen AC, Shirayama Y, Shin KH, Neve RL, et al.
Hsieh H, Boehm J, Sato C, Iwatsubo T, et al. (2006).
(2001). Expression of the cAMP response element
AMPAR removal underlies Abeta-induced synaptic
binding protein (CREB) in hippocampus produces
depression and dendritic spine loss. Neuron 52, 831–843.
an antidepressant effect. Biological Psychiatry 49,
Ji Y, Pang PT, Feng L, Lu B (2005). Cyclic AMP controls
BDNF-induced TrkB phosphorylation and dendritic spine
Chen F, Madsen TM, Wegener G, Nyengaard JR (2008a).
formation in mature hippocampal neurons. Nature
Changes in rat hippocampal CA1 synapses following
Neuroscience 8, 164–172.
imipramine treatment. Hippocampus 18, 631–639.
Karege F, Vaudan G, Schwald M, Perroud N, et al. (2005).
Chen Y, Dube CM, Rice CJ, Baram TZ (2008b). Rapid loss
Neurotrophin levels in postmortem brains of suicide
of dendritic spines after stress involves derangement of
victims and the effects of antemortem diagnosis and
spine dynamics by corticotropin-releasing hormone.
psychotropic drugs. Molecular Brain Research 136, 29–37.
Journal of Neuroscience 28, 2903–2911.
Kelleher IIIrd RJ, Govindarajan A, Tonegawa S (2004).
Chen Y, Rex CS, Rice CJ, Dube CM, et al. (2010). Correlated
Translational regulatory mechanisms in persistent forms
memory defects and hippocampal dendritic spine loss
of synaptic plasticity. Neuron 44, 59–73.
after acute stress involve corticotropin-releasing hormone
Kim Y, Sung JY, Ceglia I, Lee KW, et al. (2006).
signaling. Proceedings of the National Academy of Sciences
Phosphorylation of WAVE1 regulates actin polymerization
USA 107, 13123–13128.
and dendritic spine morphology. Nature 442, 814–817.
Cheung ZH, Fu AK, Ip NY (2006). Synaptic roles of
Knobloch M, Mansuy IM (2008). Dendritic spine loss and
Cdk5 : implications in higher cognitive functions and
synaptic alterations in Alzheimer's disease. Molecular
neurodegenerative diseases. Neuron 50, 13–18.
Neurobiology 37, 73–82.
Deng W, Aimone JB, Gage FH (2010). New neurons and new
Kobayashi S, Ishiguro K, Omori A, Takamatsu M, et al.
memories : how does adult hippocampal neurogenesis
(1993). A cdc2-related kinase PSSALRE/cdk5 is
affect learning and memory? Nature Reviews Neuroscience
homologous with the 30 kDa subunit of tau protein
11, 339–350.
kinase II, a proline-directed protein kinase associated
Dillon C, Goda Y (2005). The actin cytoskeleton : integrating
with microtubule. FEBS Letters 335, 171–175.
form and function at the synapse. Annual Review of
Korte M, Carroll P, Wolf E, Brem G, et al. (1995).
Neuroscience 28, 25–55.
Hippocampal long-term potentiation is impaired in mice
Ethell IM, Pasquale EB (2005). Molecular mechanisms of
lacking brain-derived neurotrophic factor. Proceedings of
dendritic spine development and remodeling. Progress
the National Academy of Sciences USA 92, 8856–8860.
in Neurobiology 75, 161–205.
Krishnan V, Nestler EJ (2008). The molecular neurobiology
Fischer A, Sananbenesi F, Pang PT, Lu B, et al. (2005).
of depression. Nature 455, 894–902.
Opposing roles of transient and prolonged expression of
Kurup P, Zhang Y, Xu J, Venkitaramani DV, et al. (2010).
p25 in synaptic plasticity and hippocampus-dependent
Abeta-mediated NMDA receptor endocytosis in
memory. Neuron 48, 825–838.
Alzheimer's disease involves ubiquitination of the
Fu WY, Chen Y, Sahin M, Zhao XS, et al. (2007). Cdk5
tyrosine phosphatase STEP61. Journal of Neuroscience 30,
regulates EphA4-mediated dendritic spine retraction
through an ephexin1-dependent mechanism. Nature
Lacor PN, Buniel MC, Furlow PW, Clemente AS, et al.
Neuroscience 10, 67–76.
(2007). Abeta oligomer-induced aberrations in synapse
Hains AB, Vu MA, Maciejewski PK, van Dyck CH, et al.
composition, shape, and density provide a molecular basis
(2009). Inhibition of protein kinase C signaling protects
for loss of connectivity in Alzheimer's disease. Journal of
prefrontal cortex dendritic spines and cognition from the
Neuroscience 27, 796–807.
effects of chronic stress. Proceedings of the National Academy
Lai KO, Ip NY (2009). Recent advances in understanding
of Sciences USA 106, 17957–17962.
the roles of Cdk5 in synaptic plasticity. Biochimica et
Hajszan T, MacLusky NJ, Leranth C (2005). Short-term
Biophysica Acta 1792, 741–745.
treatment with the antidepressant fluoxetine triggers
Lau CG, Zukin RS (2007). NMDA receptor trafficking in
pyramidal dendritic spine synapse formation in rat
synaptic plasticity and neuropsychiatric disorders. Nature
hippocampus. European Journal of Neuroscience 21,
Reviews Neuroscience 8, 413–426.
Li BS, Sun MK, Zhang L, Takahashi S, et al. (2001).
Hanley JG (2008). AMPA receptor trafficking pathways and
Regulation of NMDA receptors by cyclin-dependent
links to dendritic spine morphogenesis. Cellular Adhesion
kinase-5. Proceedings of the National Academy of Sciences
and Migration 2, 276–282.
USA 98, 12742–12747.
Developing cognitive enhancers from basic research
Lipton SA (2006). Paradigm shift in neuroprotection by
neurons through a phosphatidylinositol-3 kinase-
NMDA receptor blockade : memantine and beyond.
dependent pathway. Journal of Neuroscience 20, 3165–3174.
Nature Reviews Drug Discovery 5, 160–170.
Rohe M, Synowitz M, Glass R, Paul SM, et al. (2009). Brain-
Liston C, Miller MM, Goldwater DS, Radley JJ, et al.
derived neurotrophic factor reduces amyloidogenic
(2006). Stress-induced alterations in prefrontal cortical
processing through control of SORLA gene expression.
dendritic morphology predict selective impairments in
Journal of Neuroscience 29, 15472–15478.
perceptual attentional set-shifting. Journal of Neuroscience
Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, et al.
26, 7870–7874.
(2007). Extracellular interactions between GluR2 and
Lu B, Pang PT, Woo NH (2005). The yin and yang of
N-cadherin in spine regulation. Neuron 54, 461–477.
neurotrophin action. Nature Reviews Neuroscience 6,
Schindowski K, Belarbi K, Buee L (2008). Neurotrophic
factors in Alzheimer's disease : role of axonal transport.
Maekawa M, Namba T, Suzuki E, Yuasa S, et al. (2009).
Genes, Brain and Behavior 7 (Suppl. 1), 43–56.
NMDA receptor antagonist memantine promotes cell
Selkoe DJ (2002). Alzheimer's disease is a synaptic failure.
proliferation and production of mature granule neurons
Science 298, 789–791.
in the adult hippocampus. Neuroscience Research 63,
Shankar GM, Bloodgood BL, Townsend M, Walsh DM,
et al. (2007). Natural oligomers of the Alzheimer
Massa SM, Yang T, Xie Y, Shi J, et al. (2010). Small molecule
amyloid-beta protein induce reversible synapse loss
BDNF mimetics activate TrkB signaling and prevent
by modulating an NMDA-type glutamate receptor-
neuronal degeneration in rodents. Journal of Clinical
dependent signaling pathway. Journal of Neuroscience 27,
investigation 120, 1774–1785.
Matrone C, Ciotti MT, Mercanti D, Marolda R, et al. (2008).
Silva-Gomez AB, Rojas D, Juarez I, Flores G (2003).
NGF and BDNF signaling control amyloidogenic route and
Decreased dendritic spine density on prefrontal cortical
Abeta production in hippocampal neurons. Proceedings of
and hippocampal pyramidal neurons in postweaning
the National Academy of Sciences USA 105, 13139–13144.
social isolation rats. Brain Research 983, 128–136.
Monti B, Contestabile A (2009). Memory-enhancing drugs : a
Snyder EM, Nong Y, Almeida CG, Paul S, et al. (2005).
molecular perspective. Mini-Reviews in Medicinal Chemistry
Regulation of NMDA receptor trafficking by amyloid-beta.
9, 769–781.
Nature Neuroscience 8, 1051–1058.
Nagappan G, Zaitsev E, Senatorov Jr. VV, Yang J, et al.
Soetanto A, Wilson RS, Talbot K, Un A, et al. (2010).
(2009). Control of extracellular cleavage of ProBDNF by
Association of anxiety and depression with
high frequency neuronal activity. Proceedings of the National
microtubule-associated protein 2- and synaptopodin-
Academy of Sciences USA 106, 1267–1272.
immunolabeled dendrite and spine densities in
Parsons CG, Stoffler A, Danysz W (2007). Memantine : a
hippocampal CA3 of older humans. Archives of General
NMDA receptor antagonist that improves memory by
Psychiatry 67, 448–457.
restoration of homeostasis in the glutamatergic
Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, et al.
system – too little activation is bad, too much is even
(2005). Dendritic spine abnormalities in amyloid precursor
worse. Neuropharmacology 53, 699–723.
protein transgenic mice demonstrated by gene transfer and
Passafaro M, Nakagawa T, Sala C, Sheng M (2003).
intravital multiphoton microscopy. Journal of Neuroscience
Induction of dendritic spines by an extracellular domain
25, 7278–7287.
of AMPA receptor subunit GluR2. Nature 424, 677–681.
Tada T, Sheng M (2006). Molecular mechanisms of dendritic
Patterson SL, Abel T, Deuel TA, Martin KC, et al. (1996).
spine morphogenesis. Current Opinion in Neurobiology 16,
Recombinant BDNF rescues deficits in basal synaptic
transmission and hippocampal LTP in BDNF knockout
Tanaka J, Horiike Y, Matsuzaki M, Miyazaki T, et al. (2008).
mice. Neuron 16, 1137–1145.
Protein synthesis and neurotrophin-dependent structural
Pittenger C, Duman RS (2008). Stress, depression, and
plasticity of single dendritic spines. Science 319, 1683–1687.
neuroplasticity : a convergence of mechanisms.
Thome J, Sakai N, Shin K, Steffen C, et al. (2000). cAMP
Neuropsychopharmacology 33, 88–109.
response element-mediated gene transcription is
Radley JJ, Rocher AB, Miller M, Janssen WG, et al. (2006).
upregulated by chronic antidepressant treatment. Journal of
Repeated stress induces dendritic spine loss in the rat
Neuroscience 20, 4030–4036.
medial prefrontal cortex. Cerebral Cortex 16, 313–320.
Tongiorgi E, Righi M, Cattaneo A (1997). Activity-
Rakofsky JJ, Holtzheimer PE, Nemeroff CB (2009).
dependent dendritic targeting of BDNF and TrkB mRNAs
Emerging targets for antidepressant therapies. Current
in hippocampal neurons. Journal of Neuroscience 17,
Opinion in Chemical Biology 13, 291–302.
Rammes G, Danysz W, Parsons CG (2008).
Wasling P, Daborg J, Riebe I, Andersson M, et al. (2009).
Pharmacodynamics of memantine : an update. Current
Synaptic retrogenesis and amyloid-beta in Alzheimer's
Neuropharmacology 6, 55–78.
disease. Journal of Alzheimer's Disease 16, 1–14.
Righi M, Tongiorgi E, Cattaneo A (2000). Brain-derived
Waterhouse EG, Xu B (2009). New insights into the role of
neurotrophic factor (BDNF) induces dendritic targeting
brain-derived neurotrophic factor in synaptic plasticity.
of BDNF and tyrosine kinase B mRNAs in hippocampal
Molecular and Cellular Neuroscience 42, 81–89.
Z. H. Cheung and N. Y. Ip
Wei W, Nguyen LN, Kessels HW, Hagiwara H, et al. (2010).
Zakzanis KK, Leach L, Kaplan E (1998). On the nature
Amyloid beta from axons and dendrites reduces local
and pattern of neurocognitive function in major
spine number and plasticity. Nature Neuroscience 13,
depressive disorder. Neuropsychiatry, Neuropsychology,
and Behavioral Neurology 11, 111–119.
Wen Y, Yu WH, Maloney B, Bailey J, et al. (2008).
Zhang S, Edelmann L, Liu J, Crandall JE, et al. (2008).
Transcriptional regulation of beta-secretase by p25/cdk5
Cdk5 regulates the phosphorylation of tyrosine 1472
leads to enhanced amyloidogenic processing. Neuron 57,
NR2B and the surface expression of NMDA receptors.
Journal of Neuroscience 28, 415–424.
Woo NH, Teng HK, Siao CJ, Chiaruttini C, et al. (2005).
Zhou Q, Homma KJ, Poo MM (2004). Shrinkage of
Activation of p75NTR by proBDNF facilitates
dendritic spines associated with long-term
hippocampal long-term depression. Nature
depression of hippocampal synapses. Neuron 44,
Neuroscience 8, 1069–1077.
Source: http://repository.ust.hk/ir/bitstream/1783.1-8094/1/S1461145710001537a.pdf
MD Research News Tuesday February 8 , 2011 This free weekly bulletin lists the latest published research articles on macular degeneration (MD) as indexed in the NCBI, PubMed (Medline) and Entrez (GenBank) databases. These articles were identified by a search using the key term "macular degeneration". If you have not already subscribed, please email Rob Cummins at [email protected] with ‘Subscribe to MD Research News' in the subject line, and your name and address in the body of the email.
Hospital and Health Care Sub-committee on economics and planning FOREWORD During its regular meetings the Sub-Committee on Economics and Planning of the Standing Committee of the Hospitals of the European Union (HOPE) deals with the developments regarding the health care systems of the member states. As a result of this the Sub-Committee delivered reports to HOPE's