Levitra enthält Vardenafil, das eine kürzere Wirkdauer als Tadalafil hat, dafür aber schnell einsetzt. Männer, die diskret bestellen möchten, suchen häufig nach levitra kaufen ohne rezept. Dabei spielt die rechtliche Lage in der Schweiz eine wichtige Rolle.
Microsoft word - sebbar-mcs-2011.doc
Chia Laguna, Cagliari, Sardinia, Italy, September 11-15, 2011
REACTIVITY, THERMOCHEMISTRY AND KINETICS
of 2-BUTANONE RADICALS:
2 C(=O)CHCH3, CH3C(=O)CH•
CH3 and CH3C(=O)CH2CH2
N. Sebbar*, J. W. Bozzelli** and H. Bockhorn*
[email protected]
*KIT- Karlsruhe Institut of Technology, Engler-Bunte-Institut, Verbrennungstechnik
Engler-Bunte Ring 1, D-76131 Karlsruhe, Germany
** Department of Chemical Engineering, Chemistry and Environmental Science,
New Jersey Institute of Technology, Newark NJ 07102 USA
Abstract
Thermochemistry and reaction paths for the radicals of 2-butanone and for species resulting
from the reactions of 2-butanone-1yl, -3yl and -4yl radicals with 3O2 are reported. Standard
enthalpies, bond energies and kinetic parameters are evaluated using ab initio (G3MP2B3 and G3), and Density Functional (B3LYP/6-311g(d,p)) calculation. The CH2•C(=O)CH2CH3, CH3C(=O)CH•CH3 and CH3C(=O)CH2CH2• radicals + O2 association results in chemically
activated peroxy radicals with 27, 26 and 35 kcal mol-1 excess of energy, respectively. The chemically activated adducts can react back to butanone-yl + O2, form cyclic ethers, eliminate
HO2 to form an olefinic ketone, or undergo rearrangement via intramolecular abstraction of
hydrogen to form hydroperoxide-alkyls. The hydroperoxide-alkyl radical intermediates can
undergo further reactions forming cyclic ethers ketones (lactones) and OH radicals. Quantum
RRK analysis is used to calculate k
(E) and master equation analysis is used for evaluation of
pressure fall-off in these chemical activated reaction systems.
Introduction
Ketones are important in the chemistry of the atmosphere and in combustion systems.
Their photo-dissociation in the lower atmosphere results in formation of free radicals and that influence the oxidation capacity of the atmosphere. Ketones are also used as fuel tracers for monitoring fuel properties such as concentration, temperature, density, pressure, velocity, and distribution using laser-induced fluorescence [1, 2] and as fuel additives in reducing soot emissions [3,4]. Important atmospheric and combustion loss processes for ketones involve hydrogen abstractions by OH radicals, a process that is partially controlled by carbon-hydrogen bond energies, and by photolysis resulting from absorption by the carbonyl group [5, 6, 7]. The photolysis is reported to account for significant overall global presence of OH and HO2 radicals particularly in the upper troposphere [8, 9]. To our knowledge, there are no
studies describing the thermochemistry of these carbonyl alkyl radicals or the effect of the carbonyl group on the radical association reactions with O2 in atmospheric or combustion
systems. The peroxy radicals, resulting from the association of the radicals with oxygen (Figure 1), can undergo propagation and/or chain branching reactions through their chemical activated (energized)) and stabilized adducts. In this study the C—H and C—OO bond energies are determined for the three carbons of the 2-butanone and the respective peroxy radicals. The thermochemistry of reaction intermediates and the oxidation reaction paths of the 2-butanone 1-, -3- and 4- yl radicals (CH2•C(=O)CH2CH3, CH3C(=O)CH•CH3 and CH3C(=O)CH2CH2•) with dioxygen is also
determined. Structures and enthalpies of formation for important stable species and intermediate radicals resulting from this association reaction; reaction paths, transition state barriers and kinetics are reported. Standard enthalpies,
, are calculated using ab initio
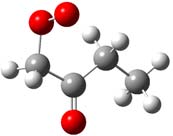
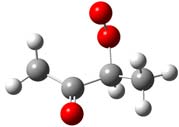
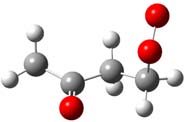
and Density Functional calculations. Kinetics and intermediate paths are determined as a function of temperature and pressure using bimolecular chemical activation analysis. CH2OO•C(=O)CH2CH3 CH3C(=O)CHOO•CH3 CH3C(=O)CH2CH2OO•
System-1 System-2 System-3
Figure 1. Structure of the three peroxy radicals
Computational Methods
Molecular properties are calculated by using the Gaussian 03 program suite [10, 11, 12].
The hybrid DFT method B3LYP, which combines the three parameter Becke exchange functional, B3, with the Lee-Yang-Parr non-local correlation functional, LYP, with a double polarized basis, 6-311G(d,p) [13, 14, 15], the G3 and the modified G3 methods reported as G3MP2B3 which uses B3LYP geometries (G3MP2//B3LYP/6-31G(d,p)) [16, 17].
The accuracy of Density Functional Theory can be improved by use of isodesmic
reactions, in which a hypothetical reaction that has the same number and type of bonds on both side, is used to calculate the enthalpy of the reaction (
). Here the errors on a
given type of bond on each side of the reaction will cancel, and the resulting accuracy of
will be high. The enthalpy of formation of a species is determined as follows:
= Σ (total energies at 298 K of products) – Σ (total energies at 298 K of reactants)
= Σ (experimental
of products) – Σ (experimental
A majority of the calculations in this study are based on isodesmic reactions, in which
calculated values are coupled to experimental or computed enthalpies of formation of the
reference species. In developing the isodesmic work reactions, efforts were made to preserve
the bonding environment in the target species, so as to effectively cancel errors in bond
energies and adjacent interaction energies across the work reactions.
Transition states were identified by their single imaginary frequency, whose mode of
vibration connects the reactants and products. Enthalpies of formation on low energy
geometries were calculated from isodesmic reaction analysis. For transition states, enthalpies
of formation were obtained from the computational enthalpy of activation and from the
calculated enthalpies of formation of the reactants or products.
Results and Discussion
Enthalpy of formation,
Standard enthalpies of formation for reactants, transition states and products of the radical oxidation reaction systems CH2•C(=O)CH2CH3, CH3C(=O)CH•CH3 and CH3C(=O)CH2CH2•
+ O2 are calculated using the total energies combined with isodesmic work reactions. The
calculated enthalpy of formation for adducts, intermediates, products, and transition state structures are given in Table 1 and in the potential diagrams below (Figures 2 - 7). Some reference species, which could not be found in the literature, were also calculated. When comparing the enthalpy values of the radicals listed in Table 1, we note an excellent agreement among B3LYP, G3MP2B3 and G3, with a difference ≤ 1 kcal mol-1.
Table 1. Calculated Standard Enthalpy of formation
B3LYP G3MP2B3 G3
CH2•C(=O)CH2CH3
-12.80 -13.57 -13.45
CH3C(=O)CH•CH3
-19.24 -18.83 -18.17
CH3C(=O)CH2CH2•
-8.54 -8.47 -8.58
CH2OO•C(=O)CH2CH3
-41.42 -40.18 -40.52
CH3C(=O)CHOO•CH3
-44.95 -44.24 -44.78
-44.1 -43.4 -43.7
The enthalpy values of the radicals and stable species reported in Figures 2 to 7, also show
good agreement among the calculation methods as further developed and reported in other
studies [18, 19]. The entropy and heat capacity for intermediates, transition state structures
and final products are calculated using the rigid-rotor-harmonic-oscillator approximation [20,
21, 22] and based on the calculated parameters: frequencies, moments of inertia from the
optimized lowest energy geometries, symmetry, spin degeneracy and optical isomers. These
values are available from the authors [19]. The thermodynamic properties have been used for
the determination of the reaction path energies and kinetic parameters in the kinetics.
Bond dissociation energies
The bond energies (BE) of C—H in the 2-butanone allow the estimation of abstraction kinetic
parameters via rate constant rules using thermochemistry. The C—H bond energies for the
CH3C(=O)CH2CH3 are determined by DFT and ab-initio (G3MP2B3, G3) calculations. Table
2 shows an excellent agreement between the DFT and the ab initio BE values. Comparison of
the C—H bonds in the 2-butanone, shows that bond energies are subject to a strong influence
of the carbonyl group. As shown in previous studies [23, 24] and in Table 2, the sp2 structure
with π bonding of the carbon oxygen double bond, allows for resonance with the adjacent
carbon radical resulting in lower bond energies. Only carbon 4 (C-4) shows a conventional
C—H BE for a primary carbon of 100.6 kcal mol-1. Table 2 shows that the C—H bond in the
secondary carbon (C-3) adjacent to the carbonyl (–C=O) in CH3C(=O)CH(—H)CH3 (2-
butanone-3yl) has the lowest bond energy, 90 kcal mol-1. The BE increases by near 6 kcal
mol-1 for the primary carbon (C-1) in (H—)CH2C(=O)CH2CH3 (2-butanone-1yl) and by 10
kcal mol-1 in the primary carbon (C-4), CH3C(=O)CH2CH2(—H) (2-butanone-4yl) which is
separated from the carbonyl by an sp3 CH2 group.
Table 2. Calculated C—H and R—OO bond energies in kcal mol-1
96.32 95.55 95.67
89.88 90.29 90.95
System-3 CH3C(=O)CH2CH2(—H) 100.58
System-1 (•OO—)CH2C(=O)CH2CH3
28.62 26.61 27.07
System-2 CH3C(=O)CH(—OO•)CH3
25.71 25.41 26.61
System-3 CH3C(=O)CH2CH2(—OO•)
35.56 34.93 35.12
Table 2 compares the C—OO bonds in the peroxy radicals and we observe that the carbonyl group influences these C—OO bond energies as well. The results show a C—OO BE of 25 to 26 kcal mol-1 when the peroxy group is attached to the carbon adjacent to the carbonyl group
(systems 1 and 2) while the C—OO BE in 2-butanone-4-yl (system 3) is higher by near 10 kcal mol-1. The same result was observed in the benzoyl + O2 system [23].
Reaction Paths and Potential Diagrams
In this study, the major reaction pathways for oxidation of CH2•C(=O)CH2CH3,
CH3C(=O)CH•CH3 and CH3C(=O)CH2CH2• radicals are determined. A detailed kinetic
evaluation of the three systems is reported in a further study [19]. Reactions available to the energized adducts include: a- Formation of a stable peroxy radical (Figure 2) b- Reverse reaction back to butanone-yl radical + O2 (no reaction)
c- Intramolecular abstraction of H from each (three) carbons by the peroxy site (Figures 3-6). d- Peroxy radical addition at the carbonyl carbon forming a cyclic peroxide ring (Figure 7). e- RO—O bond cleavage, (only important at very high T, not included in this study). 1. Formation of peroxy radical
The association reaction of the 2-butanone radical with O2 results is in formation of an
activated peroxy radical. 2-butanone has three radical sites for the addition of O2. The reaction forms the activated peroxy [CH2OO•C(=O)CH2CH3]#, [CH3C(=O)CHOO•CH3]# and [CH3C(=O)CH2CH2OO•]# intermediates with no barrier and a relatively loose transition state. In a previous study [23], we have determined the PES for the dissociation of C6H5C(=O)OO• to C6H5C•(=O) + O2 by calculating the structure of C6H5C(=O)OO• at different C—OO
distances. Our DFT calculations did not show a saddle point for the benzoyl system and we reproduce the potential of the phenyl radical + O2 association / phenylperoxy dissociation
reaction [25, 26].
CH2 C CH2 CH3 + O
CH3 C CH CH3 + O2
CH2 C CH2 CH3
CH3 C CH CH3
Figure 2. Well depth of the 2-butanone-yl + O2 association reaction,
Two of the peroxy intermediates CH2OO•C(=O)CH2CH3 and CH3C(=O)CHOO•CH3 have a
fairly shallow well depth of 25-27 kcal mol-1 at 298K (due to loss of resonance with the carbonyl group), relative to G3 calculations as shown in Figure 2. The CH3C(=O)CH2CH2OO• radical has a significantly deeper chemical activation well at 35 kcal
mol-1 which provides added energy for further reaction of peroxy radical. This deeper well is explained by the primary radical, which has no resonance stabilization. These results are interesting since the bond energies are showing that the 2-butanone-4-yl is the less reactive radical. As reported above CH3C(=O)CH2CH2• is formed after breaking over 100 kcal mol-1
C—H bond energy while the other radicals need some 90 and 95 kcal mol-1. The Kinetic analysis below shows that the temperature plays an important role in the kinetics.
2. Intramolecular hydrogen abstraction via a 4-member ring.
All three systems will undergo intramolecular hydrogen abstractions which consist of transferring a hydrogen atom from the primary (C-1 and C-4) or secondary (C-3 adjacent to the carbonyl group) carbon to the peroxy oxygen site via a 4-member, 5-member, 6-member and 7-member ring structure. These reactions are described in Figures 3 to 6 which also show further reactions available to these adducts and compare the energetics of the three systems. Figure 3 describes the hydrogen shift from the carbon of the peroxy group (ipso carbon) to the peroxy oxygen site via a strained 4-member ring structure. These ipso radicals CH•(OOH)C(=O)CH2CH3, CH3C(=O)C•(OOH)CH3 and CH3C(=O)CH2CH•(OOH) formed
for system 1, 2 and 3 respectively, are unstable. They radicals dissociate immediately, because the weak RO—OH bond (45 kcal mol-1) cleaves allowing the strong C=O bond to form at 80 kcal mol-1 via e rearrangement. These adducts eliminate an OH and form: CH3CH2C(=O)CH=O, CH3C(=O)C(=O)CH3 and CH3C(=O)CH2CH=O for 2-butanone -1yl, -
3yl and -4yl respectively.
CH3 C CH CH3
-20 -13.5
CH2 C CH2 CH3
CH3 C CH CH3 -44.9
CH3 C CH CH
Figure 3. Intramolecular H atom abstraction / OH elimination reaction; Barrier strongly
dependent on C—H bond energy
For all three systems the overall reaction is exothermic and releases some 22 to 25 kcal mol-1 relative to the stable peroxy radicals CH2(OO•)C(=O)CH2CH3, CH3C(=O)CH(OO•)CH3 and CH3C(=O)CH2CH2(OO•). However, these abstractions have high barriers, above the entrance
channel, at 41, 38 and 45 kcal mol-1 respectively. The high barriers and shallow chemical activation well limit the importance of this reaction path at low temperature, but the importance increases at higher temperatures. 3. Intramolecular hydrogen abstraction via a strained 5 member ring.
An intramolecular abstraction of a H atom via a strained 5-member ring structure is a possible channel for CH3C(=O)CH(OO•)CH3 and CH3C(=O)CH2CH2OO• (Figure 4). This abstraction
transfers a hydrogen atom from the primary carbon (C-4) or from the secondary carbon (C-3) (adjacent to the C=O group) to the peroxy oxygen radical and forms an alkyl radical: CH3C(=O)CH(OOH)CH2• for 2-butanone-3yl and CH3C(=O)CH•CH2OOH for 2-butanone-
4yl. The 2-butanone-3yl system with 26 kcal mol-1 well needs to overcome about 33 kcal mol-
1 barrier while 2-butanone-4yl system has a lower barrier of 27 and 35 kcal mol-1 well. This low barrier for a 5 member ring H transfer results from the low C—H bond energy of the C-3
carbon. The barrier for the H-shift reaction in CH3C(=O)CH•CH2OOH at -17 kcal mol-1 is 9 kcal mol-1 under the entrance channel while the reaction of CH3C(=O)CH(OO•)CH3 to CH3C(=O)CH(OOH)CH2• is 6 kcal mol-1 above the entrance channel. The 2-butanone-4yl
shows a clear energetic advantage to forward reaction (oxidation) relative to the 2-butanone -1yl or -3yl radicals.
CH3 C CH2 CH2
CH3 C CH CH2
3 C CH2 CH2
Figure 4. H-shift reaction via 5-member ring
4. Intramolecular hydrogen abstraction via a 6-member ring.
A second intramolecular abstraction via a less strained 6-member ring structure is possible for 2-butanone-1-yl, CH2OO•C(=O)CH2CH3 on the secondary C-3 (system 1) carbon and for 2-butanone-3yl, CH3C(=O)CHOO•CH3 and on the primary C-1 carbon (system 2) as illustrated
CH2 C CH2 CH3
CH3 C CH CH3 + O2
CH2 C CH CH3
CH2 C CH CH3
CH2 C CH2 CH3
Figure 5. H-shift reaction via 6-member ring
It results in the formation of a resonantly stabilized hydroperoxide radical, CH2(OOH)C(=O)CH•CH3, formed at -38 kcal mol-1 at a barrier slightly below the entrance channel by 4 kcal mol-1. The CH2•C(=O)CH(OOH)CH3 hydroperoxide alkyl radical formed
at -35 kcal mol-1, resulting from 2-butanone-3yl, has a barrier that is at the same energetic level as the entrance channel at -17.7 kcal mol-1. 5. Intramolecular hydrogen abstraction via a 7-member ring.
Abstraction of a hydrogen atom via a 7-member ring is possible from the primary carbons (C-4) in 2-butanone-1-yl and in 2-butanone-4yl forming CH2(OOH)C(=O)CH2CH2• and from CH2•C(=O)CH2CH2(OOH) respectively (Figure 6). To abstract the hydrogen from the
primary carbons (C-1 and C-4) barriers and energies show that systems 1 and 3 involve some
25-26 kcal mol-1 relative to the stable peroxy. The barrier for 2-butanone-1-yl is 26.5 at -14.7 kcal mol-1, 1 kcal mol-1 below the entrance channel while in 2-butanone-4-yl, the TST structure is at -17.5 kcal mol-1 which is 9 kcal mol-1 below the entrance channel. This difference is explained by the C—H bond energy. As shown in Table 2, in 2-butanone-1-yl, the H to extract has a C—H bond which is 4 kcal mol-1 stronger than in 2-butanone-4-yl.
CH2 CH3 + O2
CH2 C CH2 CH2
CH2 C CH2 CH2
2 C CH2 CH3
CH3 C CH CH
Figure 6. H-shift reaction via 7-member ring
6. Addition of the peroxy-oxygen radical to the C=O carbonyl group carbon C-2 Addition of the peroxy oxygen radical site on the carbonyl group is endothermic relative to the R• + O2 entrance channel energy (Figure 7). The addition to the carbonyl involves a
barrier of 33 to 38 kcal mol-1 relative to the peroxy radical as it breaks the strong (80 kcal mol-1) carbonyl π bond. This addition forms a reactive dioxetane radical with a tight transition state for 2-butanone -1yl and 3-yl systems. This dioxetane radicals, when formed dissociates
CH3 C CH CH3
CH3 C CH CH3
CH2 C CH2 CH3
CH3 C CH2 CH2
Figure 7. Addition to the carbonyl group carbon
rapidly through a very low or no barrier. Addition of the peroxy oxygen on the carbonyl group carbon in 2-butanone-4yl incurs the same barrier but with the deeper well, the barrier is near the entrance channel (R• + O2) which is at -8.5 kcal mol-1. In addition, the formation of a
reduced strain, 5- member ring, results in the formation of a less reactive radical.
Kinetic calculations on the three systems
High Pressure limit kinetic parameters are obtained from canonical Transition State Theory
calculations. Multifrequency Quantum Rice-Ramsperger-Kassel (QRRK) [27] analysis is used
to calculate k(E) data and master equation analysis (CHEMASTER code) is applied for fall-
off on the CH2•C(=O)CH2CH3, CH3C(=O)CH•CH3 and CH3C(=O)CH2CH2• + O2, reaction
systems. These form the energized peroxy radical intermediates [CH2OO•C(=O)CH2CH3]#, [CH3C(=O)CHOO•CH3]# and [CH3C(=O)CH2CH2OO•]# which undergo further reaction. The
calculations provide sets of rate constants for the formation of the stabilized adducts or reaction products as a function of pressure and temperature. Pressure dependence is important because different pressures are experienced throughout reactions in many combustion and engine systems. For example, scram jet engines run at 0.3 atm, turbines run at 10 to 15 atm, internal combustion engines can experience pressures to 100 atm and higher pressures are suggested for improved efficiency. These kinetic parameters are preliminary values and several channels are undergoing further analysis to increase accuracy.
C.C(=O)CC + O2
CO O.C(=O )C C
CO O H C(=O )C .C
C.C (= O )CC + O 2
CO O H C(=O )C C.
O=CC(=O)CC + OH
CC(YC O.CO O )
O =C C (=O )CC + O H
1000/T (1/K)
log P(ATM)
Figure 8: Chemical activation reaction for the products as a function of temperature at
P=1atm and as a function of pressure at T = 900K for CH2•C(=O)CH2CH3 + O2
Figure 8 illustrates the chemical activation reaction of CH2•C(=O)CH2CH3 + O2 system as
functions of temperature and of pressure. The results show that the important forward reactions paths are the stabilization reactions to the peroxy radical CH2OO•C(=O)CH2CH3. The reverse reaction back to CH2•C(=O)CH2CH3 + O2 is also Fast. The H-shift reaction from
the secondary carbon (C-3) shows the same dominance as the reverse reaction at low temperature; it decreases slightly at higher Temperature. The 7–member ring hydrogen abstraction reaction on the fourth primary carbon to form CH2OOHC(=O)CH2CH2• have also
some importance. Two channels start to show importance at higher temperature, at which sufficient energy is available for the energized radical to attack the oxygen site on the peroxy group carbon. This intermediate is unstable and beta scissions of an OH to form O=CHC(=O)CH2CH3 occurs.
CC(=O)CO O.C
CC(=O)C.C + O 2
CC(=O)C(=O )C + O H
CC(=O)C=C + HO2
C C (= O )C O O .C
C C (= O )C .C + O 2
C Y (C2 O 2 O .C )
C .C (=O )C O O H C
C C (= O )C O O H C .
C C (= O )C (=O )C + O H
C C (= O )C = C + HO 2
100 0 /T (1 /K )
log P(ATM )
Figure 9: Chemical activation reaction for the products as a function of temperature at
P=1atm and as a function of pressure at T = 900K for CH3C(=O)CH•CH3 + O2
The chemical activation reaction of CH3C(=O)CH•CH3 + O2 system illustrated in Figure 9,
shows that the important forward reaction paths are the stabilization reactions to the CH3C(=O)CHOO•CH3 peroxy radical. The next two important channels are the OOH
molecular elimination form CH3C(=O)CH=CH2 and the hydrogen abstraction reaction on the first primary carbon to form CH2•C(=O)CHOOHCH3. More minor product a few orders of
magnitude slower, are the butanedione + OH, the hydrogen abstraction from the primary carbon (C-4) and the oxygen attack on the carbonyl group carbon. These three reactions are in competition at higher temperature.
CC(=O)CC. + O2
CC(=O)CC. + O2
CC(=O)CC(=O) + OH
CC(=O)CC(=O) + OH
CC(=O)C=C + HO2
CC(=O)C=C + HO2
1000/T (1/K)
log P(ATM)
Figure 10: Chemical activation reaction for the products as a function of temperature at
P=1atm for CH3C(=O)CH2CH2• + O2
Figure 10 illustrates the product profiles from the 2-butanone-4yl peroxy radical, CH3C(=O)CH2CH2•, reaction with O2 versus temperature at 1 atm and pressure at 900K. The reaction to stabilization of the CH3C(=O)CH2CH2OO• peroxy radicals, is the most important
forward reaction at low temperature. Two new product channels show importance also: intramolecular H-abstraction from C-3 to form CH3C(=O)CH•CH2OOH and molecular
elimination of HO2 forming CH3C(=O)CH=CH2. Abstraction of H-atom from C-1 is lower in
magnitude but still of importance. The formation of the butadione + OH from the ipso
abstraction channel is markedly increased with increasing temperature and is an important
path at high temperature. The channel of less importance is as expected the attack of the
oxygen radical on the carbonyl group carbon breaking the 80 kcal mol-1 C=O double bond.
Conclusions
Enthalpy of important intermediates, transition state structures and products resulting from
the reaction of 2 butanone carbon radicals: CH2•C(=O)CH2CH3, CH3C(=O)CH•CH3 and CH3C(=O)CH2CH2• radicals with O2, were studied by using DFT (B3LYP/6-311G(d,p) level)
and ab-initio (G3MP2B3 and G3). These are illustrated in the PE diagrams of this study and are also reported separately in tabular form [19]. The R• + O2 association of the three systems
results in chemically activated peroxy radicals with 26 kcal mol-1 for 2-butanone-1yl and 2-butanone-3yl and with 35 kcal mol-1 for the 2-butanone-4yl radical. Important forward reaction channels of these chemically activated radical are: stabilization to peroxy radicals, OH and HO2 plus olefin and intramolecular paths. Ether (lactone) ring formation occurs
subsequent to the intramolecular hydrogen abstraction reactions. Stabilization to the peroxy radical is the most important channel.
References
[1]
Schulz, C.; Sick, V. Prog. Energy Combust. Sci. 31: 7 (2005)
Hanson, R. K.; Seitzman, J. M.; Paul, P. H. Appl.Phys. B 50: 441 (1990)
Pepiot-Desjardins, P.; Pitsch, H.; Malhotra, R.; Kirby, S. R.; Boehman, A. L. Combust. Flame 154: 191 (2008)
Hong, Z.; Davidson, D. F.; Vasu, S. S.; Hanson, R. K. Fuel 88: 1901 (2009)
Gierczak, T.; Burkholder, J. B.; Bauerle, S.; Ravishankara, A. R. Chem. Phys.,231, 229 (1998)
Blitz, M. A.; Heard, D. E.; Pilling, M. J. J. Phys. Chem. A, 110, 6742 (2006)
J. Phys. Chem. 95, 10816 (1991)
Singh, H. B.; Kanakidou, M.; Crutzen, P. J.; Jacob, D. J. Nature 378, 50 (1995)
Wennberg, P. O.; Hanisco, T. F.; Jaeglé, L.; Jacob, D. J.; Hintsa, E. J.; Lanzendorf, E. J.; Anderson, J.G.; Gao, R. S.; Keim, E. R.; Donnelly, S. G.; Del Negro, L. A.; Fahey, D. W.; McKeen, S. A.; Salawitch, R. J.; Webster, C. R.; May, R. D.; Herman, R. L.; Proffitt, M. H.; Margitan, J. J.; Atlas, E. L.; Schauffler, S. M.; Flocke, F.; McElroy, C. T.; Bui, T. P. Science, 279, 49 (1998)
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Zakrzewski, V. G., Montgomery, J. A., Jr., Stratmann, R. E., Burant, J. C., Dapprich, S., Millam, J. M., Daniels, A. D., Kudin, K. N., Strain, M. C., Farkas, O., Tomasi, J., Barone, V., Cossi, M., Cammi, R., Mennucci, B., Pomelli, C., Adamo, C., Clifford, S., Ochterski, J., Petersson, G. A., Ayala, P. Y., Cui, Q., Morokuma, K., Salvador, P., Dannenberg, J. J., Malick, D. K., Rabuck, A. D., Raghavachari, K., Foresman, J. B., Cioslowski, J., Ortiz, J. V., Baboul, A. G., Stefanov, B. B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Gomperts, R., Martin, R. L., Fox, D. J., Keith, T., Al-Laham, M. A., Peng, C. Y., Nanayakkara, A., Challacombe, M., Gill, P. M. W., Johnson, B., Chen, W., Wong, M. W., Andres, J. L., Gonzalez, C., Head-Gordon, M., Replogle, E. S., and Pople, J. A., Gaussian 98, Gaussian, Inc., Pittsburgh PA, (Revision A.11), 2001.
[11] http://www.gaussian.com/index.htm. [12]
Foresman, J. B., Frisch., A., "Exploring chemistry with electronic structure methods : a guide to using Gaussian", - 1. ed., Pittsburg, Pa.: Gaussian, 1993.
Becke, A. D. J. Chem. Phys. 98, 5648, (1993).
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 37, 785, (1988).
Montgomery, J. A.; Ocherski, J. W.; Petersson, G. A., J. Chem. Phys. 101, 5900, (1994).
Redfern, P. C.; Zapol, P.; Curtiss, L. A and Raghavachari, K. J. Phys. Chem. A, 104: 5850, (2000).
[17] Baboul, A. G.; Curtiss, L. A.; Redfern, P. C.; Raghavachari, K. J. Chem. Phys. A, 110:
(16),7650-7657, (1999)
[18] Sebbar, N.; Bockhorn, H.; Bozzelli, J. W., proceeding of the European combustion
Symposium, Cardiff, 2011.
[19] Sebbar, N.; Bockhorn, H.; Bozzelli, J. W., manuscript in preparation. [20] Sheng, C. Ph D Dissertation, Department of Chemical Engineering, Chemistry and
Environmental Science, New Jersey Institute of Technology, Newark (2002)
Lay, T. H.; Krasnoperov, L. N.; Venanzi, C. A.; Bozzelli, J. W. J. Phys. Chem. 100,824 (1996)
Sebbar, N.; Bockhorn, H.; Bozzelli, J. W., Int. J. Chem Kinet 40: 583–604 (2008)
[23] Sebbar, N.; Bozzelli, J. W.; Bockhorn, H. MCS6, Ajaccio ,Corsica, France, June 7-11, 2009 [24] Sebbar, N.; Bockhorn, H.; Bozzelli, J. W., submitted to J. Phys. Chem.A. [25] Tokmakov, I. V.; Kim, G-S; Kislov, V. V.; Mebel, A. M.; M. C. Lin J. Phys.Chem. A 109,
6114-6127 (2005)
N. Sebbar, H. Bockhorn, J. W. Bozzelli Combust. Sci. and Tech., 180: 959–974, (2008)
[27] Sheng, C.; Bozzelli, J. W.; Dean, A. M.; Chang, A. Y. J. Phys.Chem. A,106,7276-7293 (2002)
Source: http://www.combustion-institute.it/proceedings/MCS-7/papers/RKC/RKC-03.pdf
International Bulletin of Pharmaceutical Sciences 2012, 1 (1-2): 17–29 International Bulletin of Pharmaceutical Sciences Article Type: Minireview Use of Metabolomics for Monitoring Metabolic Responses Caused by Drug Administration 2. Application of Metabolomics in the Study of Metabolic Changes Induced by Drugs
Eur Arch Otorhinolaryngol (2002) 259 : 279–284DOI 10.1007/s00405-002-0456-z L.Guarda-Nardini · R. Tito · A. Staffieri · A. Beltrame Treatment of patients with arthrosis of the temporomandibular joint by infiltration of sodium hyaluronate: a preliminary study Received: 20 June 2001 / Accepted: 25 January 2002 / Published online: 24 April 2002 © Springer-Verlag 2002