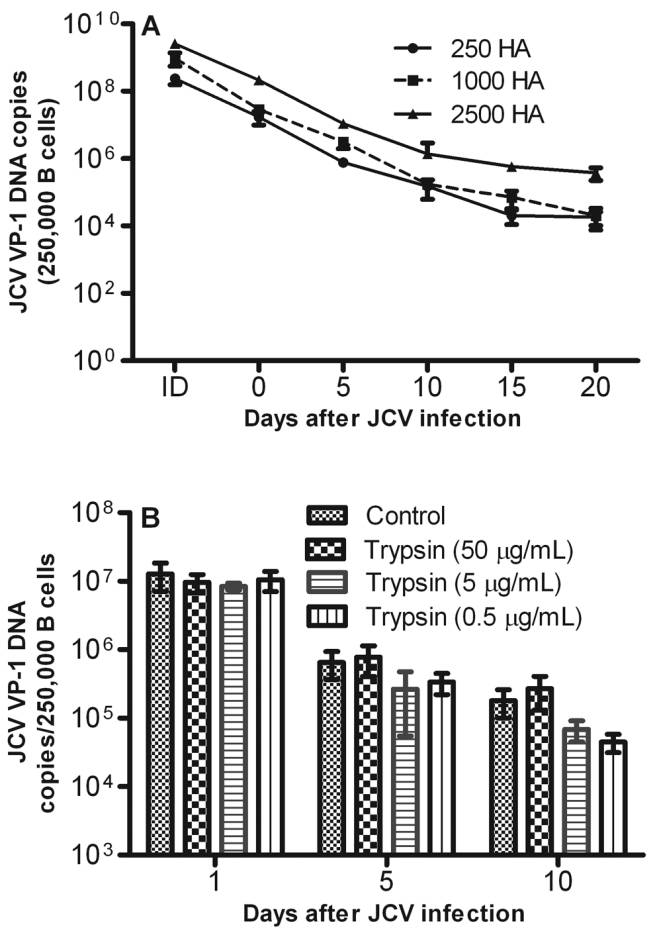
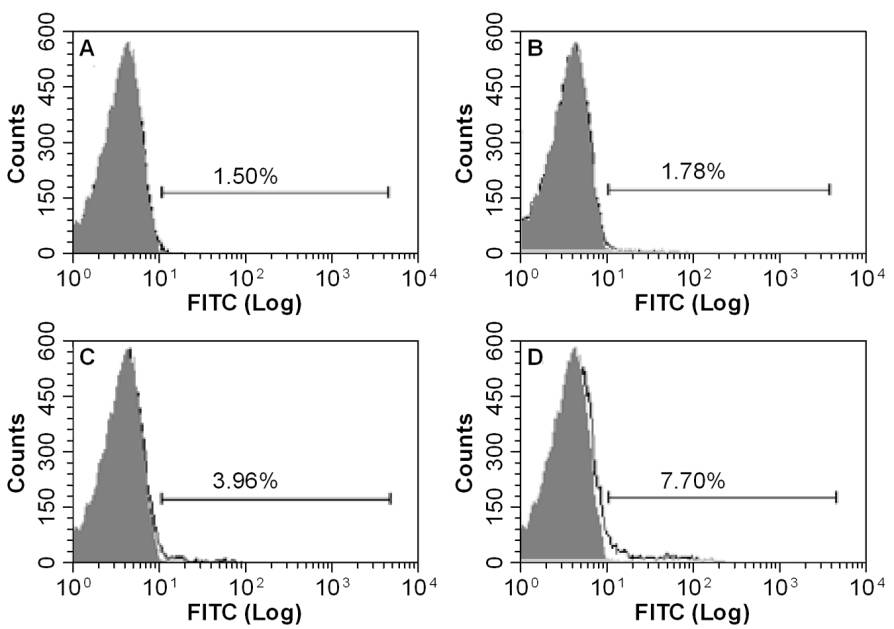
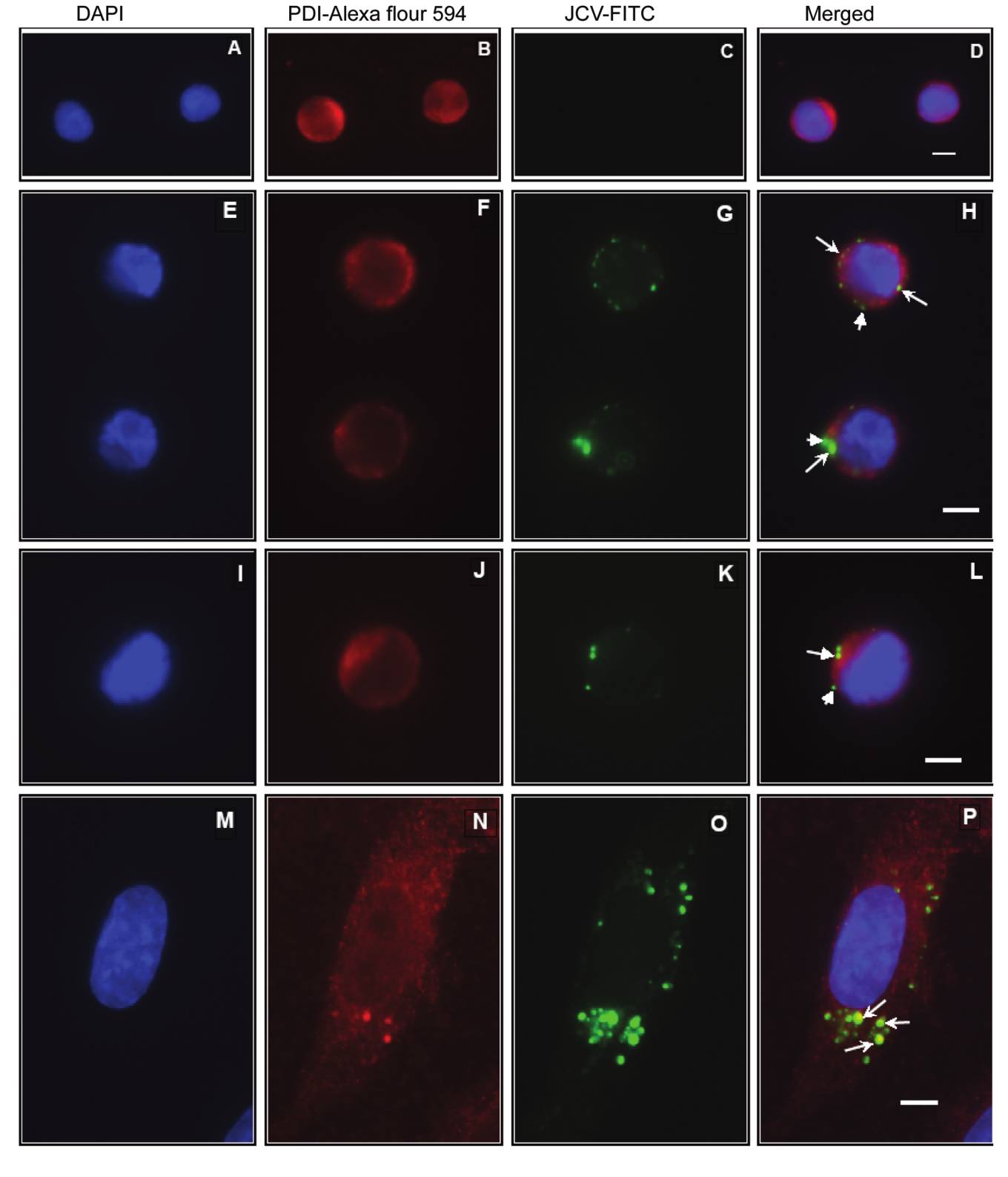
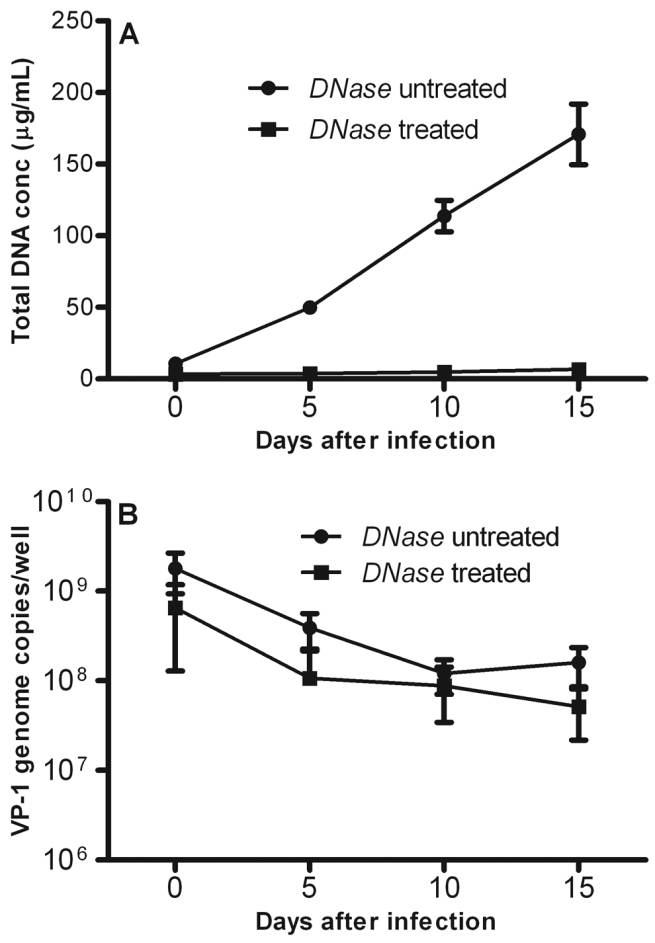
sbts.edu
& the Authority of Christ by Russell D. Moore Counseling and the Authority of Christ: A New Vision for Biblical Counseling at The Southern Baptist Theological Seminary "And they were astonished at His teaching, for He taught them as one having authority, and not as the scribes." [Mark 1:22 ESV] The story of The Southern Baptist Theological Seminary is seen most clearly not in