Levitra enthält Vardenafil, das eine kürzere Wirkdauer als Tadalafil hat, dafür aber schnell einsetzt. Männer, die diskret bestellen möchten, suchen häufig nach levitra kaufen ohne rezept. Dabei spielt die rechtliche Lage in der Schweiz eine wichtige Rolle.
Main.dvi
EN CIENCIAS E INGENIERÍAS
Estudio computacional B3LYP de la interacción del hidrógeno molecular [H2
] con rccc
R-Pyg[4]arenos [R = metil, flúor] funcionalizados con Li+
Andrés S. Urbina 1
, Andrea A. Saltos1
, F. Javier Torres1,2∗
1
Colegio de Ciencias e Ingeniería - El Politécnico, Universidad San Francisco de Quito
Diego de Robles y Vía Interoceánica, Quito, Ecuador.
2
Grupo Ecuatoriano para el Estudio Experimental y Teórico de Nanaosistemas –GETNano–
Diego de Robles y Vía Interoceánica, Universidad San Francisco de Quito.
∗
Autor principal/Corresponding author, e-mail: [email protected]
Editado por/Edited by: Robert Cazar, M.Sc.
Recibido/Received: 11/15/2011. Aceptado/Accepted: 11/27/2011.
Publicado en línea/Published on Web: 12/20/2011. Impreso/Printed: 12/20/2011.
The interaction of molecular hydrogen [H2] with methyl- and fluoride-substituted
rccc pi-rogallol[4]arenes functionalized with Li+ cations [Li-R-Pyg[4]Ar] was theoretically stu-died by means of DFT quantum-mechanical calculations at the B3LYP/6-311G(d,p) levelof theory. In a first stage of the study, the stability of the Li+ cation within the cavityof the R-Pyg[4]arenes was analyzed by inspecting the local environment of the adsorbedion and the total density as well as the electrostatic potential maps of the complexes. Ina subsequent stage of the work, the optimal position of a H2 molecule in the cavity ofthe pure and lithium-functionalized R-Pyg[4]arenes was determined. Upon obtaining theequilibrium geometry, the BSSE-corrected binding energy of the various H2/R-Pyg[4]Arcomplexes was calculated. The results showed that the sorption capacity of R-Pyg[4]arenesis significantly improved by the presence of the Li+ cation within their cavity.
Keywords. Hydrogen storage, pyrogallol, DFT, sorption capacity.
La interacción del hidrógeno molecular [H2] con
rccc metil- y fluor-pirogalol[a]arenosfuncionalizados con cationes Li+ [Li-R-Pyg[4]Ar] fue estudiado teóricamente por mediode cálculos cuanto-mecánicos DFT al nivel de teoría B3LYP/6-311G(d,p). En una primeraetapa de estudio, la estabilidad del catión Li+ dentro de la cavidad de los R-Pyg[4]arenosfue analizada inspeccionando el ambiente local del ion adsorbido y los mapas de densidadtotal de carga así como de potencial electrostático de los complejos. En una siguiente etapade trabajo, se determinó la posición óptima del H2 en la cavidad de los R-Pyg[4]arenospuros, y con litio. Una vez obtenida la geometría de equilibrio, la energía de amarre librede ESFB fue calculada para varios complejos H2/R-Pyg[4]Ar. Los resultados muestranque la capacidad de adsorción de los R-Pyg[4]arenos mejora de manera significativa por lapresencia del catión Li+ dentro de su cavidad.
Palabras Clave. Almacenamiento de hidrógeno, pirogalol, DFT, capacidad de adsorción.
vo aumento en el consumo de energía que se origina acausa del crecimiento poblacional y a la gran demandaenergética que han venido mostrando los países en vías
Actualmente la mayor parte de la energía primaria utili-
de desarrollo [2]. Hoy en día, la demanda de energía es
zada a nivel mundial proviene de los combustibles fósi-
de aproximadamente 5.13 x 1017 BTU, la cual se cree
les, los cuales son recursos no renovables que se extraen
que aumentará en un 49 % para el año 2035 [3]. En este
de yacimientos que cuentan con reservas limitadas. Pro-
contexto, el desarrollo de fuentes de energías alternati-
yecciones recientes estiman que las reservas mundia-
vas ha adquirido gran importancia, ya que en un futu-
les de petróleo y gas natural (
i.e. principales combus-
ro cercano deberán reemplazar a los combustibles fósi-
tibles fósiles) durarían 38 y 57 años, respectivamente, si
les en las diferentes actividades de la humanidad. Una
se mantiene el ritmo de consumo energético actual [1].
de las opciones más prometedoras es la de trasladar la
A este hecho se tiene que agregar también el progresi-
Avances, 2011, Vol. 3, No. 2, Pags. A30-A34
Urbina et al.
Avances, 2011, Vol. 3, No. 2, Pags. A30-A34
economía mundial hacia el uso de hidrógeno molecu-lar como vector energético [2]. La denominada "Econo-mía del Hidrógeno" involucra tres pasos fundamentales:producción, almacenamiento y uso [3].
Obervando que actualmente existen medios eficientespara la producción y uso de H2 como vector energético.
La transición a la economía del hidrógeno se encuen-tra frenada por las dificultades que se tiene actualmentecon respecto al proceso de almacenamiento, el cual seha convertido en un desafío tecnológico ya que el H2,en condiciones estándar de presión y temperatura, es ungas muy poco compresible [4].
En los últimos años, se han propuesto varias alterna-tivas para el almacenamiento del hidrógeno molecular,siendo una de éstas la fisisorción o adsorción en sólidosmicropporosos[5]. A este respecto, numerosos materia-les han sido investigados de forma experimental y teóri-ca como por ejemplo: carbon activado[6], nano-estructu-ras de carbono[6], metal-organic frameworks (MOFs)
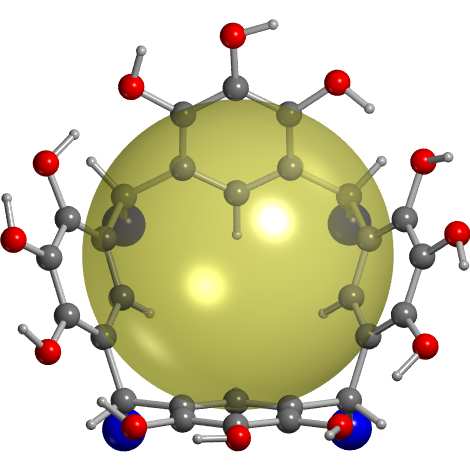
Figura 1: R-Pyg[4]Ar con conformacoón de copa rccc. La esfera
transparente de color amarillo representa el espacio libre dentro
[7], polímeros inorgánicos, zeolitas [8, 9, 10, 11], entre
de la cavidad del macrociclo. Los átomos de carbono, oxígeno e
otros; pero hasta ahora no se ha encontrado ningún ma-
hidrógeno de la estructura son representados con las esferas de
terial que cumpla con las metas técnicas sugeridas por
color gris, rojo y blanco, respectivamente. Por claridad, los gru-
pos sustituyentes R unidos al plano de la base de la copa son re-
el departamento de energía de USA (i.e. tiempos rápi-
presentados por las esferas de color azul.
dos de carga y descarga, temperatura y presión cercanasa las condiciones estándar) para usar eficientemente el
R con carácter electro-aceptor vacían el interior de la
H2 en aplicaciones móviles o estacionarias [6]. En un
cavidad [18]. Esta importante propiedad se puede apro-
reporte reciente [12], se ha determinado que para lograr
vechar para funcionalizar los R-Pyg[4]Ar con cationes,
un óptimo almacenamiento de H2, la energía de interac-
los cuales poseen la cualidad de mejorar la capacidad de
ción que debe existir entre un material adsorbente y el
adsorción de H2 como ha sido reportado en varios estu-
H2 a temperatura y presión estándar es de 15.1 kJ/mol
dios realizados sobre zeolitas intercambiadas con meta-
[12], valor que aún no ha sido alcanzado para ningún
les alcalinos (i.e. Li+, Na+ y K+) [8, 9, 11]. En dichos
estudios se ha concluido que el ion litio posee una ma-yor afinidad por el hidrógeno molecular en comparación
El presente trabajo tiene por objeto analizar mediante
a otros cationes monovalentes, especialmente en el caso
cálculos cuanto-mecánicos en modelos moleculares la
interacción del H
2CHA-5/1 [10] en donde el catión Li+ se encuentra
2 con los rccc pirogalol[4]arenos (R-
Pyg[4]Ar) funcionalizados con cationes litio; para deesta manera evaluar su potencial como medio para al-
Tomando en cuenta lo mencionado, la presente investi-
gación teórica se enfoca en el análisis de las propiedades
de adsorción del metil- y del F-Pyg[4]Ar tanto funciona-
lizados con Li+ (1 y 2) como puros (3 y 4). Consideran-
do estos cuatro sistemas, se pretende evaluar el efecto delos grupos electro-donadores y electro-aceptores sobre
la estabilidad del ion en el interior de los macrociclos, yconsecuentemente, sobre su capacidad de adsorción de
Pese a que se conoce que los R-Pyg[4]Ar pueden adop-
tar varias conformaciones [13, 14, 15, 16] (i.e. rctt: es-tructura de silla o rccc: estructura de copa), en el pre-
Para la construcción de los modelos moleculares del
sente estudio teórico, se ha considerado solamente la
metil- y del F-Pyg[4]Ar, se empleó como punto de par-
conformación de copa rccc debido a que su estructura
tida los datos de difracción de rayos X reportados pa-
ofrece el ambiente adecuado para la adsorción de molé-
ra el decil-Pyg[4]Ar el cual cristaliza en conformación
culas de pequeñas y medianas dimensiones [16] (Figu-
de copa rccc como ha sido reportado por Dueno et. al.
ra 1). Adicional a esto, un reporte reciente ha sugerido
[19]. En la macromolécula obtenida a partir de la cel-
que las propiedades electronícas de estos compuestos
da unitaria del cristal, se reemplazaron los sustituyentes
se pueden controlar variando los grupos sustituyentes R
decil por los grupos metil y flúor para obtener el Me-
en la base de su estructura[17, 18]. En términos genera-
Pyg[4]Ar y el F-Pyg[4]Ar, respectivamente. En una pri-
les, se ha observado que grupos R con carácter electro-
mera fase del estudio, se realizó la optimización geomé-
donador generan pozos electrónicos en el interior de la
trica de los modelos considerando que ambas macromo-
copa de los rccc R-Pyg[4]Ar, mientras que los grupos
léculas pertenecen al grupo punto de simetría C4. Una
Avances, 2011, Vol. 3, No. 2, Pags. A30-A34
Urbina et al.
Li-Me-Pyg[4]Ar 2.664, 2.663, 2.553, 2.664
2.612, 2.612, 2.612, 2.612
Tabla 1: Distancias entre el Li+ y los átomos de carbono
(dLi−C′s) enlazados a los grupos sustituyentes de los rccc R-
Pyg[4]Ar y ángulos diedro (α) del catión con respecto al plano
de la base en cada compuesto. La energía de interacción corregi-
da con respecto al ESFB (EIc) entre el catión y cada R-Pyg[4]Ar
es también reportada. Las distancias estan reportadas en Å, los
ángulos en grados y las energías en kJ/mol.
vez determinada la estructura de equilibrio de cada com-puesto, se añadió el catión Li+ dentro de la cavidad enel centro del plano correspondiente a la base de la es-
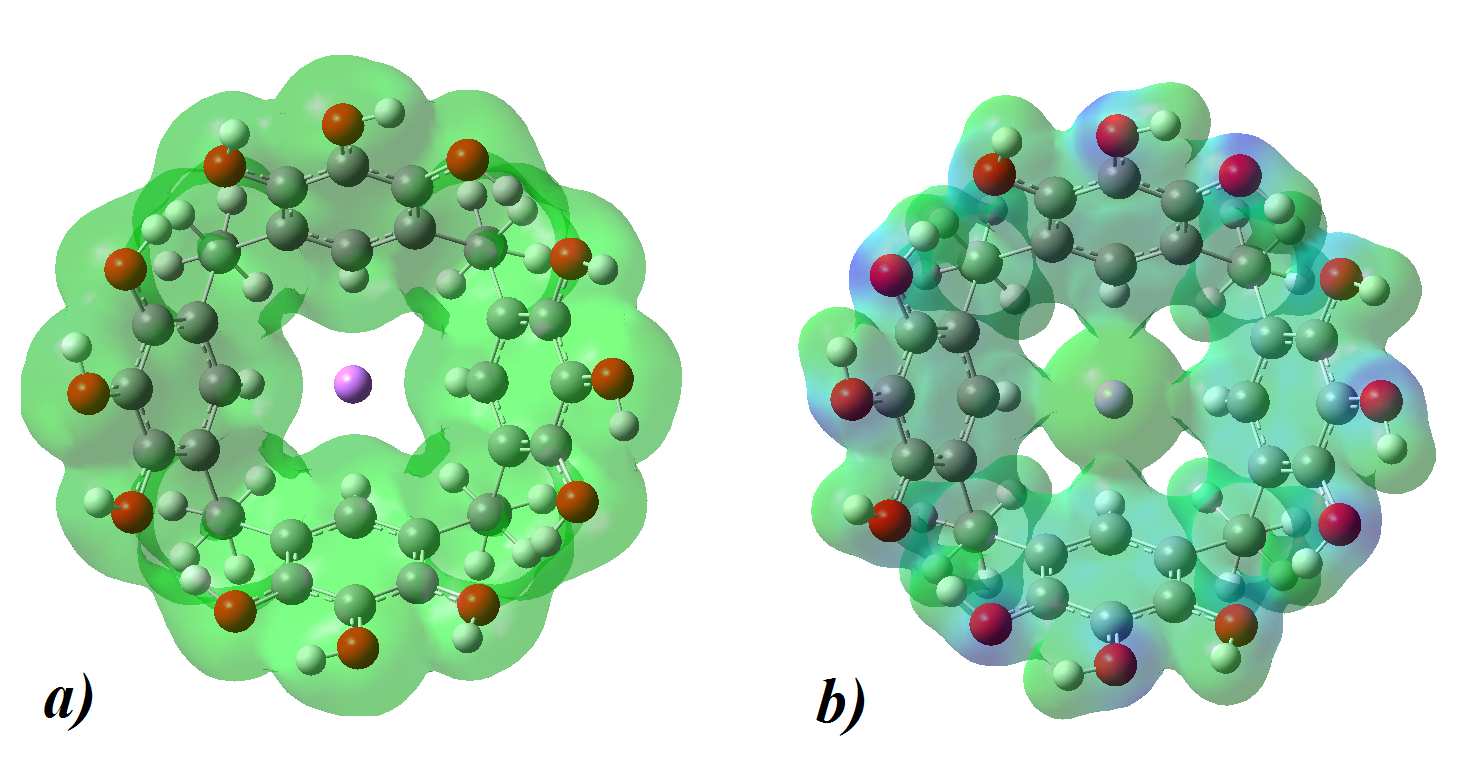
Figura 2: Mapas de densidad electrónica (a) y potencial elec-
trostático (b) del Li-Me-Pyg[4]Ar, obtenidos usando isovalores de
tructura de copa (Figura 1); y posteriormente, se deter-
0.008 a.u. y 0.2 a.u., respectivamente.
minó la posición optima del catión mediante un nuevoproceso de optimización geométrica. Las propiedades
Resultados y Discusión
electrónicas y la estabilidad del catión en el interior delMe-Pyg[4]Ar y del F-Pyg[4]Ar fueron investigadas en
Estructura de los Li-R-Pyg[4]Ar
las geometrías resultantes.
En la Tabla 1 se resumen los datos geométricos corres-pondientes al ambiente geométrico en el cual se encuen-
En una siguiente fase de la investigación, se procedió al
tra el catión Li+ en los compuestos 1 y 2. Como se pue-
análisis de la interacción de H2 con los compuestos 1-4.
de observar, para el complejo 1 la distancia promedio
La molécula de hidrógeno fue colocada perpendicular al
entre los átomos de carbono enlazados a los grupos sus-
eje de simetría de las macromoléculas cerca de la base
tituyentes R y el Li+ es de
de la estructura de copa. Posteriormente, la posición del
∼2.66Å, mientras que pa-
ra 2 la distancia promedio es
∼2.61Å. La diferencia de
2 fue relajada hasta su geometría óptima dentro de la
0.05Å entre las distancias medidas para cada caso se de-
cavidad de los compuestos 1-4, y en la geometría resul-
be a los distintos ambientes electrostáticos en los que se
tante se llevó a cabo el cálculo de la energía de amarre
encuentra el catión en cada macromolécula. En el Me-
empleando la siguiente expresión[20]:
Pyg[4]Ar, se observa que el Li+ se encuentra alejado dela base de la estructura de copa (α = 47◦) debido a que
EA[H2−Pyg] = E[H2] + E[Pyg] − E[H2−Pyg]
la cavidad del compuesto contiene un pozo de electro-nes (i.e., efecto de los grupos metilo [18]) el cual estabi-
donde el primer término de la parte derecha de la ecua-
liza al catión sin que éste pase a formar parte de la ma-
ción se refiere a la energía del hidrógeno molecular, el
cromolécula. Por el contrario en el caso del F-Pyg[4]Ar,
segundo a la energía de cada compuesto 1-4, y el terce-
el catión Li+ se acomoda de mejor manera en la base del
ro a la energía del complejo H2/Li-R-Pyg[4]Ar o H2/R-
compuesto (α = 45◦) debido a que gran parte de la car-
Pyg[4]Ar. Las energías de amarre calculadas a través de
ga del macrociclo (i.e., densidad electrónica) se localiza
la Ecn. 1 fueron posteriormente corregidas restándoles
en los átomos de flúor, los cuales son muy electronegati-
el error causado por la superposición de funciones base
vos. Esta observación es importante considerando que la
(ESFB) [21], mediante el método Boys-Bernardi [22].
capacidad para interactuar con hidrógeno molecular que
Los valores de energía corregidos se denotarán como
posee un catión monovalente presente en un determina-
do material depende en gran medida de que tan expuestose encuentra dentro de su estructura, como se ha con-
luido para la zeolita CHA intercambiada con metalesalcalinos en varios estudios teóricos [10, 11]. Siguien-
Los cálculos de optimización geométrica y de punto sim-
do esta idea, se espería entonces que el catión Li+ al
ple de energía fueron realizados con el programa Gaus-
encontrarse más expuesto dentro del Me-Pyg[4]Ar, in-
sian09 [23] empleando el funcional B3LYP (i.e., Bec-
teractúe muy fuertemente con el H2 en comparación al
ke 3-parameter hybrid exchange functionals and Lee-
Li+ presente en el interior del F-Pyg[4]Ar.
Yang-Parr correlation functionals) junto a las funcionesbase 6-311G(d,p) [24] como nivel de teoría. Los mo-
Antes de proseguir al estudio de la interacción del H2
delos moleculares fueron construidos usando el progra-
con los compuestos 1-4, se debe analizar la estabilidad
ma MOLDRAW [25] y posteriormente fueron refina-
del Li+ presente dentro del metil- y del F-Pyg[4]Ar me-
dos con el software GaussView5. Los mapas de densi-
diante el cálculo de la energía de amarre entre el catión
dad electrónica y potencial electroestático de los Li-R-
y cada macromolécula. Los valores correspondientes a
Pyg[4]Ar fueron generados a partir de sus funciones de
la energía de amarre (EAc) están reportados en la Tabla
onda por medio de la aplicación cubegen[23] asociada
1. Como se puede apreciar para ambos compuestos (1 y
a Gaussian09. Posteriormente, los mapas de densidad y
2), la interacción entre el Li+ y los R-Pyg[4]Ar es fa-
potencial de fueron visualizados con GaussView5.
vorable con valores calculados de ∼314 kJ/mol y ∼288
Urbina et al.
Avances, 2011, Vol. 3, No. 2, Pags. A30-A34
teóricas anteriores [8, 9, 10, 11] en donde se demuestra
que el H2 interactúa de manera significativa con catio-
nes casi libres presentes en la estructura de materiales
microporosos ya que estos poseen una mayor capacidad
polarizante (Figura 2b).
Tabla 2: Distancias medidas desde los puntos de referencia has-
ta la molécula H2 para los complejos estudiados. Las distancias
están reportadas en Å y las energías en kJ/mol.
kJ/mol, respectivamente. En el caso particular del com-
En el presente artículo se estudió la interacción del H2
puesto 1 se puede observar que existe una interacción
dentro de los Li-R-pyg[4]arenos con sustituyentes metil
mayor en comparación al compuesto 2, lo que señala
y flúor, mediante cálculos cuanto-mecánicos a un nivel
una mayor estabilidad del Li+ en el interior del Me-
de teoría B3LYP/6-311G(d,p). Los resultados muestran
Pyg[4]Ar, hecho que se observa también al analizar los
que la energía de amarre entre el H2 y los R-Pyg[4]Ar
mapas de densidad y potencial electroestático de este
incremente cuando las macromoléculas se encuentran
compuesto. En la Figuras 2a y 2b, los mapas de den-
funcionalizadas con el catión Li+. Se ha observado ade-
sidad electrónica y potencial electrostático, respectiva-
más que la capacidad de polarización del catión dismi-
mente, muestran como la carga negativa en la base de la
nuye en presencia de sustituyentes con mayor electro-
copa Me-Pyg[4]Ar estabiliza al catión Li+ a la vez que
negatividad, por lo que el catión Li+ interactua fuer-
lo deja expuesto (Figura 2a) permitiendo que el mismo
temente con H2 cuando se encuentra en el interior de
no pierda su capacidad polarizante (Figura 2b).
un R-Pyg[4]Ar con sustituyentes electrodonadores co-mo el grupo metil. Analizando las energías de amarre de
Interacción del H2 con R-Pyg[4]Ar y Li-R-Pyg[4]Ar
8.72 y 8.06 kJ/mol obtenidas para los complejos H2/Li-Me-Pyg[4]Ar y H2/Li-F-Pyg[4]Ar, respectivamente, se
En la Tabla 2 se resumen los datos referentes a la po-
puede concluir que estos macrocompuestos funciona-
sición resultante del H2 en los compuestos 1-4 después
lizados con cationes tienen un gran potencial para ser
del proceso de optimización geométrica en cada caso.
usados como medio para adsorber hidrógeno molecular
La posición del H2 fue determinada midiendo la distan-
en comparación a otros materiales previamente estudia-
cia desde un punto de referencia en la cavidad de cada
dos como las zeolitas [9, 10] debido a que los valores
uno de las estructuras hasta el centro de masa de la mo-
calculados de EAc se acercan al límite de 15.1 kJ/mol
lécula (DH2−Ref). Para los compuestos funcionalizados
establecido como óptimo para aplicaciones móbiles y
con Li+, el punto de referencia establecido fue la posi-
ción del catión; mientras que para los compuestos puros,se tomó como referencia la posición virtual en donde seencontraría el Li+.
Como se puede observar, para el compuesto 1 la dis-
Los autores agradecen a la Universidad San Francis-
co de Quito por financiar el presente estudio a través
−Ref es ∼2.07Å, mientras que para 3 ésta
distancia es ∼2.71Å. De la misma manera para 2 dicha
del programa Chancellor Grants 2011. FJT agradece
distancia es de ∼2.07Å, mientras que para 4 es ∼3.09 Å.
de forma especial a la Corporación Ecuatoriana para el
Es evidente entonces, una disminución en las distancias
Desarrollo del Internet Avanzado (CEDIA) por el co-
para en los complejos 1 y 2, lo que indica una mejor ab-
financiamiento de este trabajo y por haber apoyado a la
sorción de los R-Pyg[4]Ar cuando el átomo de litio está
creación del Grupo Ecuatoriano para el Estudio Experi-
mental y Teórico de Nanosistemas (GETNano) graciasa los recursos financieros y logísticos brindadados en el
La anterior deducción es además sustentada por los va-
marco de las convocatorias CEPRA2 y CEPRA4.
lores de las energías de amarre corregidas por el ESFB
(EAc) obtenidas para los complejos 1-4. Como está re-
portado en la Tabla 2, la energía de amarre para el com-
puesto 1 es 8.72 kJ/mol y para el compuesto 2 es 8.06
kJ/mol; en cambio, para los complejos 3 y 4 las energías
[1] Conti, J., Doman, L., Smith, K., and Mayne, L. 2010.
"International Energy Outlook 2010." U.S.E.I. Adminis-
obtenidas fueron -1.58 y -1.09 kJ/mol, respectivamente.
Estos resultados muestran que en los R-Pyg[4]Ar pu-ros la molécula de hidrógeno siente una fuerza repulsi-
[2] Turner, J. 2001. "A Relizable Renewable Energy Futu-
va que empuja la molécula fuera de la cavidad; mien-
re." Science. 414, 332–337.
tras que en el caso de los macrociclos funcionalizados
[3] Barreto, L., Makihira, A., and Riahi, K. 2003. "The Hy-
con el catión Li+ los valores positivos sugieren un esta-
drogen Economy in the 21st Century: A Sustainable De-
do estable de enlace lo que a su vez se traduce en una
velopment Scenario." Int. J. Hydrogen Energ. 28, 267–
mejora en la capacidad de adsorción de los compues-
tos. Es importante mencionar también que estos resulta-
[4] Crabtree, G., Dresselhaus, M., and Buchanan, M. 2004.
dos están en perfecta concordancia con investigaciones
"The Hydrogen Economy." Phys. Today. 57, 39–44.
Avances, 2011, Vol. 3, No. 2, Pags. A30-A34
Urbina et al.
[5] Zuttel, A. 2004. "Hydrogen Storage Methods." Natur-
[23] Frisch, M., G. W., Schlegel, H. B., Scuseria, G. E., Robb,
wissenscharten. 91, 157–172.
M. A., Cheeseman, J. R., Scalmani, G., Barone, V. Men-
[6] Annemikie, W., and Van derBerg, A. 2008. "Materials
nucci, B. P. G. A., Nakatsuji, H., Caricato, M., Li, X.,
for Hydrogen Storage: Current Research Trends and
Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G.,
Perspectives". Chem. Commun. 668–681.
Sonnenberg, J. L., Hada, M., Ehara, M. Toyota, K., Fu-kuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Hon-
[7] Zhou, L. 2005. "Problems and Progress in Hydrogen
da, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery,
Storage Methods." Renew. Sust. Energ. Rev. 9, 395–408.
J. J. A., Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd,
[8] Torres, F. J., Civalleri, B., Terentyev, A., Ugliengo, P.,
J. J., Brothers, E., Kudin, K. N. Staroverov, V. N., Ko-
and Pisani, C. 2005. "Theoretical Study of Molecular
bayashi, R., Normand, J., Raghavachari, K., Rendell, A.,
Hydrogen Adsorption in Mg-Exchanged Chabazite." J.
Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M. Re-
Chem. Phys. C 111, 2505–2513.
ga, N., Millam, N. J., Klene, M., Knox, J. E., Cross,
[9] Torres, F. J., Terentyev, A., and Ugliengo, P. 2006. "Ab
J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts,
Initio Investigation of the Interaction of H
R. Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi,
Exchanged Low-Silica Chabazites." Journal of Physics:
R. Pomelli, C., Ochterski, J. W., Martin, R. L., Moro-
Conf. Series. 117, 012012(8).
kuma, K., Zakrzewski, V. G. Voth, G. A., Salvador, P.,Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas,
[10] Torres, F. J., Civalleri, B., Ricchiardi, G., and Zecchi-
Foresman, J. B. Ortiz, J. V., Cioslowski, J., and Fox, D. J.
na, A. 2007. "Interaction of H2 with Alkali- Metal-
2011. Gaussian 09, revision A1. Gaussian, Inc. Walling-
Exchanged Zeolites: A Quantum Mechanical Study." J.
Phys. Chem. B. 111, 2505–2513.
[11] Torres, F., Vitillo, J. G., Civalleri, B., Ricchiardi,
G., and Zecchina, A. 2007. "Interaction of H2 with
mistry.III.The Role of Exact Exchange. J. Chem. Phys.
Alkali- Metal-Exchanged Zeolites: A Quantum Mecha-
98, 5648–5652.
nical Study." J. Chem. Phys. C.. 111, 2505–2513.
[25] Ugliengo, P., Borzani, G., and Viterbo, Z. 2011. Mol-
[12] Bhatia, S. 2006. "Optimum Conditions for Adsorptive
draw: Molecular Graphics on a Personal Computer.
Storage." Langmuir. 22, 1688–1700.
[13] Liam, C., Palmer, R., and Jr, J. 2005. "Hydrocarbon Bin-
ding Inside a Hexameric Pyrogallol[4]arene Capsule."Org. Lett. 7, 787–789.
[14] Shivanyuk, A., Friese, J., Döring, S., and Jr, J. 2003.
"Solvent-stabilized Molecular Capsules." J. Org. Chem.
68, 6489–6496.
[15] Shivanyuk, A., Far, A., and Jr, J. 2002. "Rigid Tetrani-
troresorcinarenes." Org. Lett. 9, 1555–1558.
[16] Gutsche, D., Zouhair, A., Böhmer, V., Harrowfield, J.,
Vicens, J., and Eds, M. 2001. "Synthesis of Calixarenesand Thiacalixarenes." Springer Netherlands. 25, 155–181.
[17] Zambrano, C. H., Manzano, C. A., Saltos, A., Dueno,
E., and Zeller, M. 2010. "Síntesis de 2,8,14,20-Tetra-n-Butilpirogalol[4]areno y Estudio Computacional Con-formacional." Avances en Ciencias e Ingenierías.
[18] Manzano, S., Dueno, E. E., Zambrano, C. H., and To-
rres, F. J. "Theoretical Evidence of Electronic Tuningwithin the Cavity of rccc Pyrogallol[4]arenes." Chem.
Phys. Lett. Submitted for publication.
[19] Dueno, E., Zambrano, C., W., S., and J.P., K.
[20] Wormer, P. Theory and applications of computational
chemistry the first forty years In K.Kim In C.E. Dykstra,G. Frenking and (ed.) G.E> Scuseria, (ed.), Forty Yearsof AB Initio Calculations on Intermolecular Forces., pp.
1047–1091.
[21] Kestner, N. 1968. "he-he Interaction in the SCFMO Ap-
proximation." J. Chem. Phys. 48, 252–257.
[22] Boys, F. and Bernardi, F. 1970. "The Calculation of
Small Molecular Interactions by the Differences of Se-parate Total Energies, Somo Procedures with ReducesErrors." Mol. Phys. 110, 553–566.
Source: http://repositorio.cedia.org.ec/bitstream/123456789/293/5/Paper_AVANCES_FJTorres.pdf
An in vitro ischemic penumbral mimic perfusate increasesNADPH oxidase-mediated superoxide production in culturedhippocampal neurons Matthew E. PameSameh S. Qingbo , J. Cameron ,Xiang Q. , Laura L. , Gabriel G. aDepartment of Pediatrics, Division of Respiratory Medicine, University of California San Diego, La Jolla, CA 92093, USAbDepartment of Anesthesiology, University of California San Diego, La Jolla, CA 92093, USAcSchool of Medicine, Department of Geriatric Medicine, University of California San Diego, La Jolla, CA 92093, USAdDepartment of Neuroscience, University of California San Diego, La Jolla, CA 92093, USAeThe Rady Children's Hospital-San Diego, San Diego, CA 92123, USA
Autorin: Martina Seifert Agentur für vernetzte Kommunikation Martina Seifert studierte Germanistik, Literaturwissenschaft und Philosophie an der Universität Bielefeld mit Abschluss zur Magistra Artium. Seit 2001 als Online Redakteurin für das Content Management und die Pressearbeit bei Geizkragen.de zuständig, veröffentlichte sie 2004 das „Geizkragen-Sparbuch" (Rowohlt Verlag GmbH, Hamburg). Daneben übernahm die Autorin von 2006 bis 2008 in freier Mitarbeit Pressearbeiten für das Affiliate Netzwerk der adbutler GmbH (seit 2008 belboon-adbutler GmbH)und betreute im Rahmen des Relaunches 2007 federführend die Textredaktion für die Webseite adbutler.de. Ebenfalls seit 2006 in freier Mitarbeit als Pressereferentin und Textredakteurin für die Online Marketing Agentur der coupling media GmbH zuständig, erstellte Martina Seifert 2009 in Eigenrecherche das Online Marketing Lexikon für die Webseite coupling-media.de mit rund 200 Begriffen aus der Branche.