Levitra enthält Vardenafil, das eine kürzere Wirkdauer als Tadalafil hat, dafür aber schnell einsetzt. Männer, die diskret bestellen möchten, suchen häufig nach levitra kaufen ohne rezept. Dabei spielt die rechtliche Lage in der Schweiz eine wichtige Rolle.
A quantitative analysis of insulin signaling in neurodegeneration
College of William and Mary
A Quantitative Analysis of Insulin Signaling in
NeurodegenerationElise M. Braatz
College of William and Mary
Follow this and additional works at:
Part of the nd the
Recommended CitationBraatz, Elise M., "A Quantitative Analysis of Insulin Signaling in Neurodegeneration" (2015).
College of William & Mary UndergraduateHonors Theses. Paper 119.
This Honors Thesis is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M Publish. It has been accepted forinclusion in College of William & Mary Undergraduate Honors Theses by an authorized administrator of W&M Publish. For more information, pleasecontact .

Abstract
This project introduces two mathematical models representing the biochemical interactions
between insulin signaling, PD, and AD. The models can be used to examine the changes that
occur over the course of the disease as well as identify which processes would be the most
effective targets for treatment. The models are mathematized using Biochemical Systems
Theory. They incorporate treatment strategies that includes several experimental drugs along
with current treatments. In the past, BST models of neurodegeneration have used Power Law
Analysis and Simulation to model the system. This work recommends the use of MATLAB
instead. MATLAB allows for more flexibility in both the model itself and in data analysis.
Previous BST analyses of neurodegeneration began treatment at disease onset. As shown in these
models, the outcomes of delayed, realistic treatment and full treatment at disease onset are
significantly different. The delayed treatment strategy is an important development in BST
modeling of neurodegeneration. It emphasizes the importance of early diagnosis, and allows for
a more accurate representation of disease and treatment interactions.
1. Introduction
PD is a neurodegenerative disorder characterized by dopaminergic neuron death in the substantia
nigra pars compacta and results in physical symptoms such as tremors, rigidity, and bradykinesia
as well as psychological symptoms such as an increased risk of dementia, depression, and
anxiety. Insulin resistance, a precursor of T2DM, is a result of a diet high in fats and sugars.
Insulin signaling pathways and PD pathogenesis have been experimentally proven to converge
(Morris et al., 2008; Morris et al., 2011), yet clinical data is inconsistent (Bosco et al., 2012). The
model includes production of ROS and RNS, p38 phosphorylation, tau protein
hyperphosphorylation, inflammation, and dopamine synthesis, degradation, and transport. This
paper presents a mathematical model that details the interactions between these processes and
how they can combine to influence neuronal death.
AD is an age-related neurodegenerative disorder affecting many brain regions, notably the
cholinergic neurons in the hippocampus, which are correlated to the cognitive decline
characteristic of AD (Kerbler et al., 2015). Insulin resistance in AD is much more accepted than
in PD. Both a lack of insulin in the CNS, similar to T1DM, and a lack of CNS insulin receptor
sensitivity, similar to T2DM, have been associated with AD, leading some to suggest that AD
should be known as "type-III diabetes" (de la Monte et al., 2014). This paper presents a model of
the pathways shared between insulin signaling impairment and AD within cholinergic neurons,
astrocytes, and microglia. The data presented focuses on changes in insulin signaling,
inflammation, ROS production, mitochondrial dysfunction, Ca2+ signaling dysregulation,
neurofibrillary tangle production, Aβ oligomerization, p38 activation, and finally cell death.
These disease simulations makes use of a modeling strategy know as BST, which was
established by Savageu in 1969 and developed further by Voit in 2000. BST uses ODEs to
model changes in reaction rates and species concentrations throughout the progression of the
disease. These models include a baseline state, which depicts a healthy cell, a disease state,
modeling a cell with PD and insulin resistance, and a treatment state, which is a disease state cell
including several treatment options. The disease state was created by activating trigger points
within the baseline model that initiated disease development, and the treatment state was created
by introducing a mixture of drugs at two times: the onset of the disease, and halfway through the
disease progression. These treatments target reactions the models show are important in
neurodegenerative pathogenesis where it converges with insulin signaling pathways.
2. Methods
2.1 Modeled Pathways in PD
This section explains several key pathways in the model, demonstrating the extent to which
insulin signaling influences the biochemistry of PD. The system was modeled using the
biochemical pathway visualization program CellDesigner.
2.1.1 Insulin Signaling and Inflammation
Insulin signaling begins with insulin binding to the insulin receptor on the cellular membrane,
which causes phosphorylation of the insulin receptor substrate (Takahashi et al., 1996). Insulin
signaling is promoted by PPARG, which is also a transcription factor for proteins important in
antioxidant scavenging and mitochondrial repair (Miranda et al., 1999; Mudo et al., 2012; Xing
et al., 2007). Insulin signaling is inhibited by the chronic inflammation present in PD. The
inflammation is a result of cytokine production in astrocytes and microglia, specifically
cytokines IL-1β, IL-6, TNFα, and MCSF (Ghosh et al., 2007; Glass et al., 2010). Overproduction
of NO promoted by nitric oxide synthase (Hunot et al., 1996), prostaglandin synthesis promoted
by COX2 (Xing et al., 2007), and NF-κB in the neuron and glia are also contributing factors.
Inflammation is decreased by the presence of PPARG (Ricote et al., 1998). This process is
outlined in Model 1.
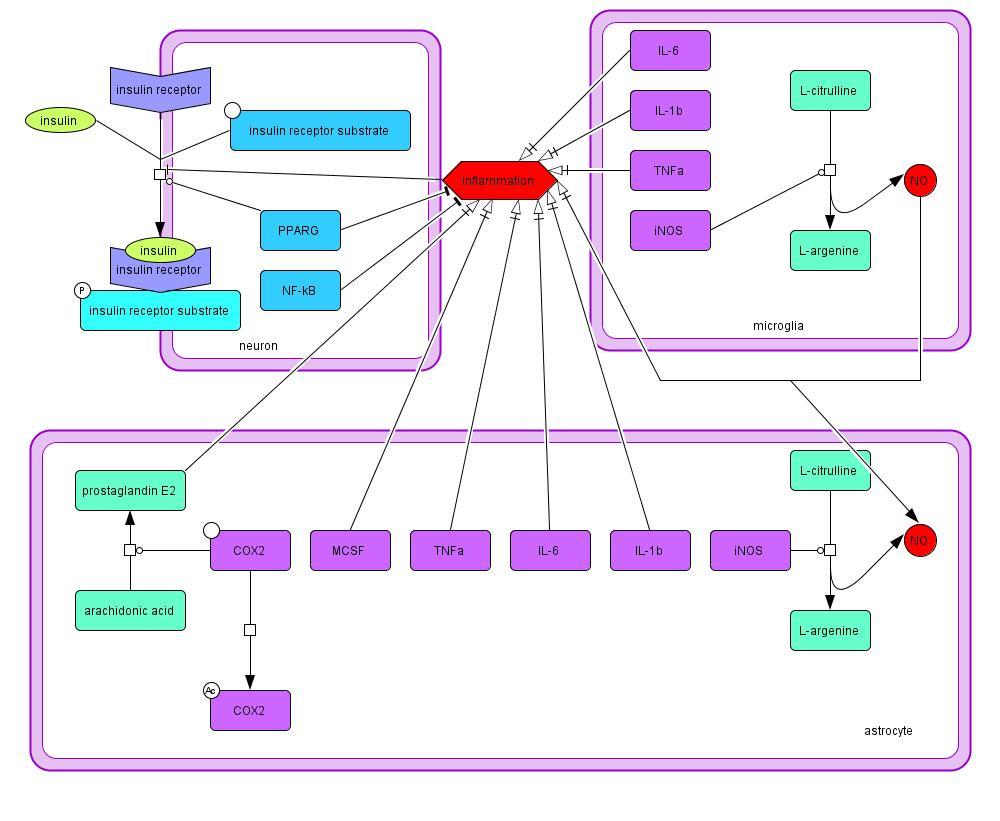
Model 1: Mechanisms of inflammation and factors influencing insulin signaling in PD
2.1.2 Dopamine
Dopamine is produced in the neuron through the conversion of tyrosine to L-DOPA, and L-
DOPA to dopamine (Model 2). Tyrosine hydroxylase, which is activated via phosphorylation by
phosphorylated p38, promotes L-DOPA synthesis, and decarboxylase promotes the final step in
the process to produce dopamine (Yu et al., 2005). Dopamine can also enter the cell through the
dopamine transporter, which is upregulated by insulin signaling (Patterson et al., 1998). Free
dopamine in the cell can be degraded into quinones, H2O2, and O2- (Yu et al., 2005), or use a
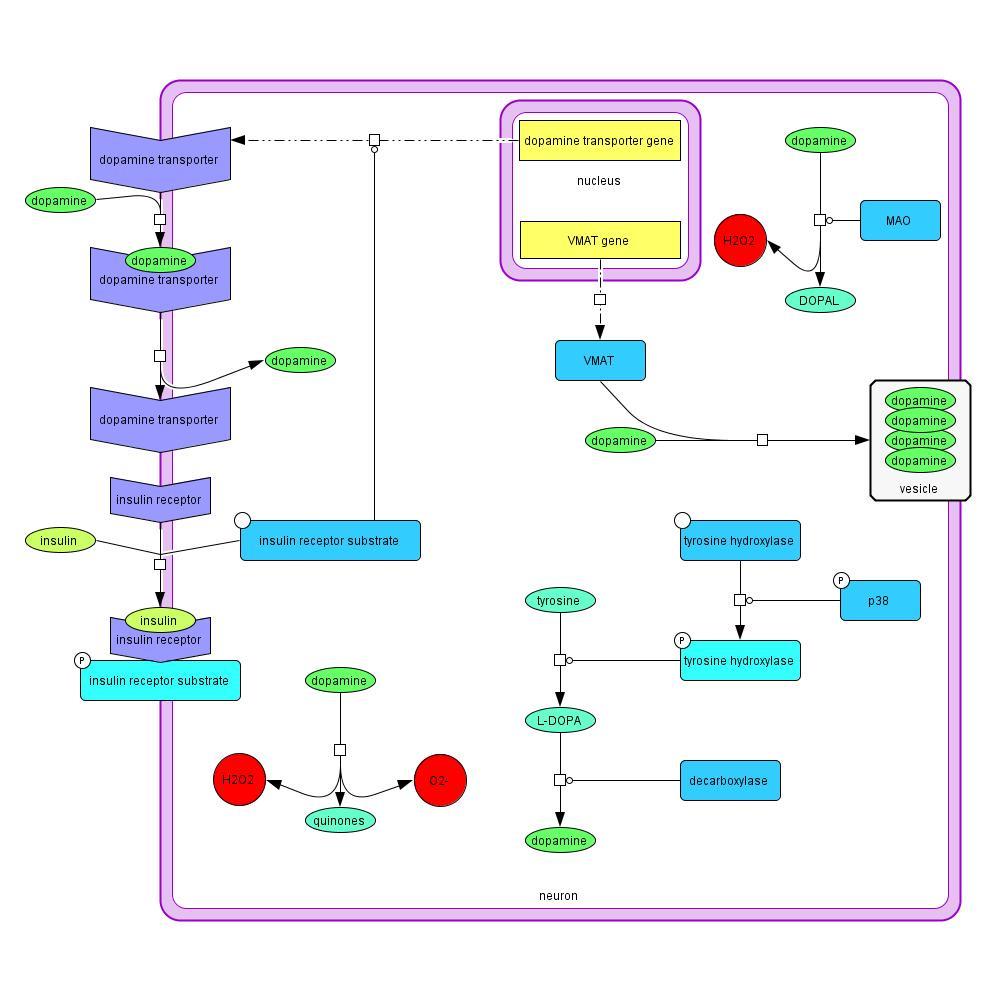
monoamine oxidase promoter, producing H2O2 and DOPAL (Marchitti et al., 2007).
Alternatively, dopamine is collected into vesicles to be released at synapses. Vesicular dopamine
is protected from degradation and the resulting production of ROS. VMAT are important in
sequestering the dopamine into vesicles. VMAT is downregulated in PD (Xiong et al., 2011).
Model 2: Reactions and cellular processes involving dopamine in PD
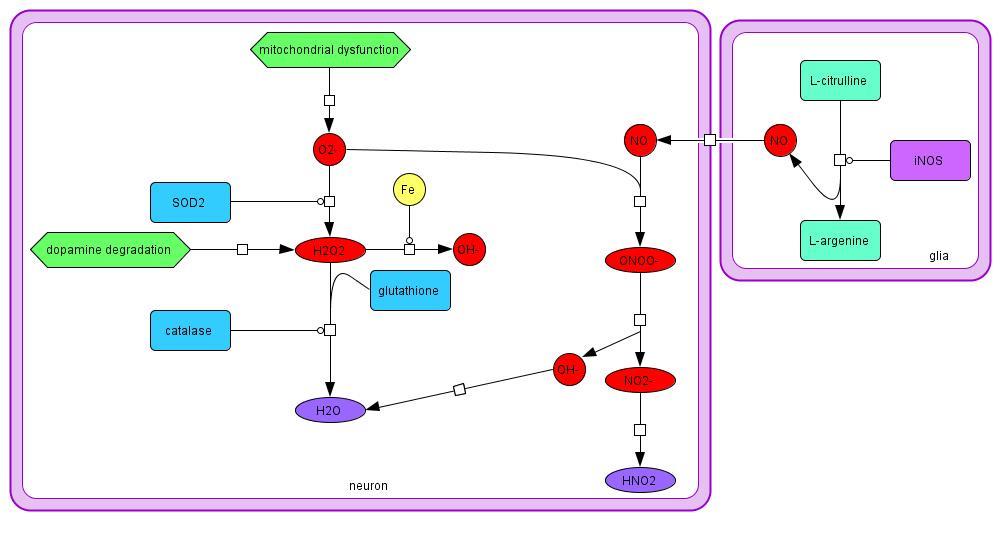
2.1.3 ROS and RNS
ROS are the result of dopamine degradation and mitochondrial dysfunction in PD. Mitochondrial
dysfunction creates an excess of O2-, which can be converted to H2O2 by SOD2. The H2O2 can
then be converted to water by glutathione or catalase (Evans et al., 2002), or OH- by Fe through
the Fenton reaction. Dopamine degradation also produces H2O2 and O2- in the cell. NO, a RNS,
is produced in both the neuron and the glia from L-citrulline, and is membrane-permeable
(Hunot et al., 1996). The NO can react with O2- to create ONOO-, which is extremely dangerous
to the neuron (Beckman, Crow, 1993). Excessive ROS and RNS can react easily with vital
cellular species causing lipid peroxidation, misfolded proteins, DNA damage, and endoplasmic
reticulum and mitochondrial stress, all of which decrease cellular function and increase the risk
of cell death. ROS and RNS can also trigger the caspase cascade, initiating apoptosis (Choi et al.,
2004; Ko et al., 2005).
Model 3: Synthesis and degradation of reactive oxygen and nitrogen species in PD
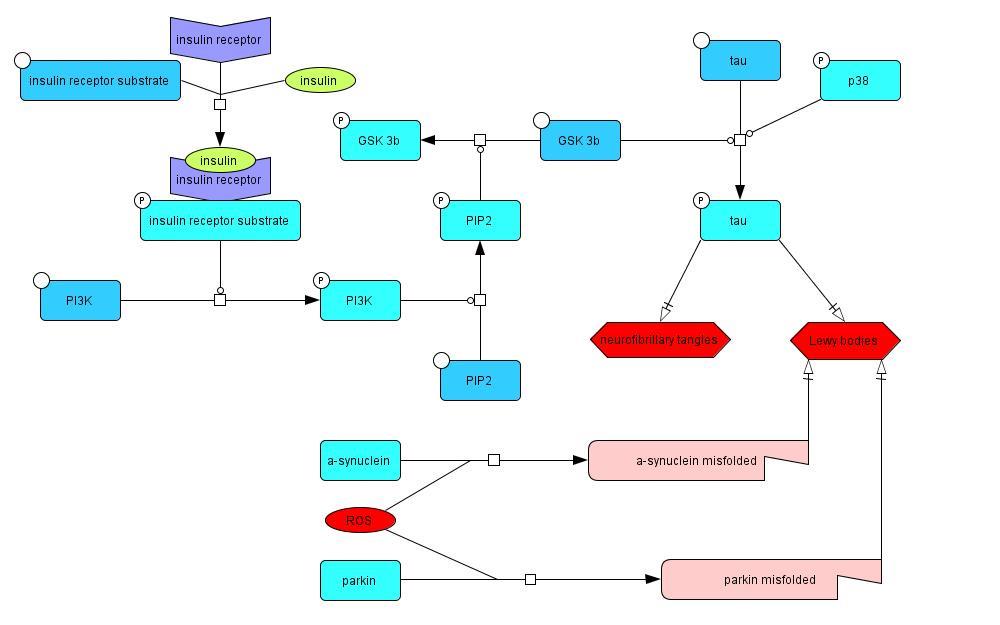
2.1.4 Tau Phosphorylation, Neurofibrillary Tangles, and Lewy Bodies
Insulin signaling phosphorylates PI3K (Piao et al., 2012), which in turn phosphorylates PIP2,
creating PIP3. PIP3 deactivates GSK-3β through phosphorylation, which prevents it from
promoting tau hyperphosphorylation (Lucas et al., 2001). Tau hyperphosphorylation is also
promoted by phosphorylated p38 (Zarubin, Han, 2005). The tau protein is important in
microtubule stabilization, so the loss of function caused by the hyperphosphorylation results in
impeded axonal transport. Hyperphosphorylated tau is a component of aggregates such as
neurofibrillary tangles and Lewy bodies, which are characteristic of PD (Arima et al., 1999;
Bancher et al., 1993). Lewy bodies also contain proteins such as parkin and α-synuclein, which
aggregate as a result of ROS interactions that cause them to misfold (Winklhofer et al, 2003;
Zheng et al., 2010).
Model 4: The effects of insulin resistance on tau hyperphosphorylation, neurofibrillary tangle
accumulation, and Lewy body production in PD
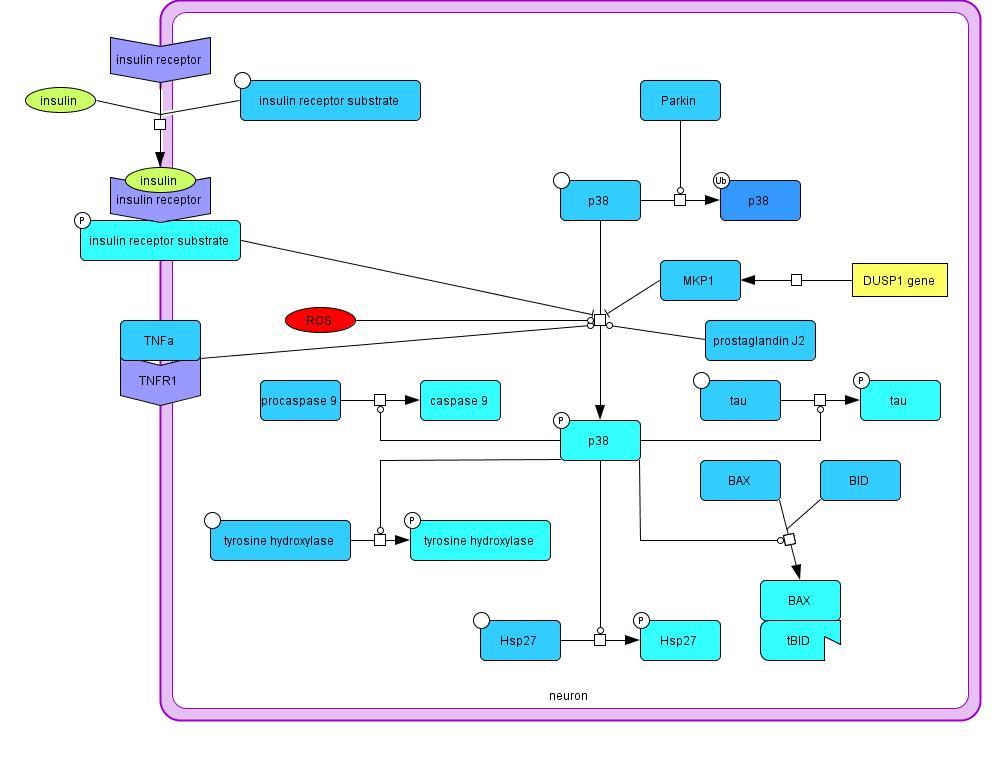
2.1.5 p38 Phosphorylation
The p38 mitogen-activated protein kinase is involved in several different pathways in this model.
p38 is activated by phosphorylation, which is promoted by prostaglandins, tumor necrosis factor
α receptor 1 signaling, and ROS (Choi et al., 2004). Inhibition of p38 phosphorylation is caused
by insulin signaling (Heidenreich, Kummer, 1996) and the MKP-1 expressed by the DUSP1
gene (Taylor et al., 2013). p38 promotes caspase 9 maturation leading to apoptosis, tau
phosphorylation, tyrosine hydroxylase activation, Bax/tBID complex formation resulting in
mitochondrial membrane depolarization and related dysfunction, and finally Hsp27 activation
Model 5: Mechanisms influencing p38 phosphorylation and its effects on the cell in PD
(Snyder et al., 2009; Yu et al., 2005; Zarubin, Han, 2005). Hsp27 can inhibit tau
phosphorylation, increase glutathione expression, inhibit Fas ligand binding, and decrease Lewy
body formation by preventing α-synuclein aggregates from accumulating in the Lewy bodies
(Choi et al., 2012; Mehlen et al., 1996; Mehlen et al., 1996a; Shimura et al., 2004; Zourlidou et
al., 2004). p38 can also be ubiquitinated by the E3 ubiquitin ligase parkin and degraded (Ko et
al., 2005). Parkin is downregulated in PD which causes an increase in p38.
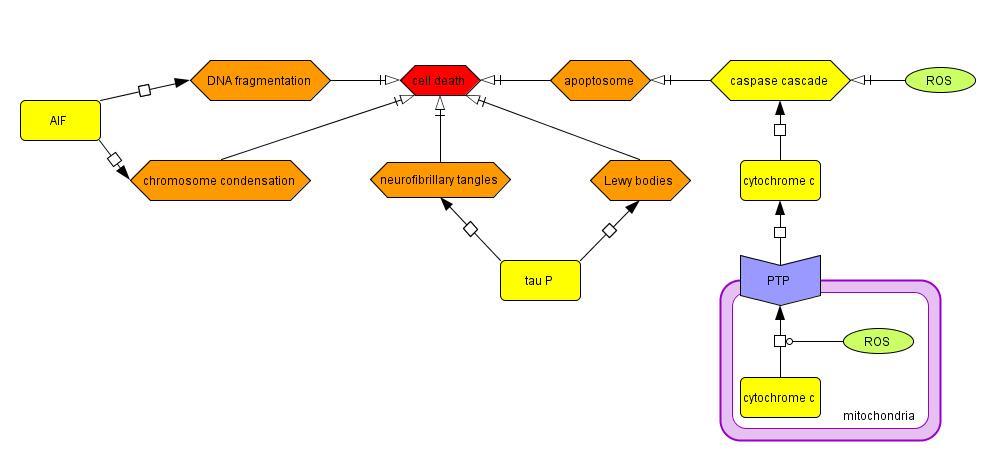
2.1.6 Cell Death
Cytosolic ROS can directly cause apoptosis by activating the caspase cascade (Choi et al., 2004;
Ko et al., 2005), and mitochondrial ROS can indirectly activate the caspase cascade by opening
the PTP of the mitochondria and releasing cytochrome c (Moon et al., 2005), SMAC/DIABLO,
and AIF. SMAC/DIABLO increases caspase activity by binding to IAP proteins, which prevent
the IAP from binding to and inhibiting caspases 3 and 9. Cell death can also be triggered in a
caspase-independent manner by DNA fragmentation and chromosome condensation initiated by
Model 6: Cellular processes contributing to the death of SNPc neurons in PD
the migration of the AIF from the mitochondria to the nucleus (Susin et al., 1999). Furthermore,
hyperphosphorylated tau can create neurofibrillary tangles and Lewy bodies, inducing cell death.
2.2 Modeled Pathways in AD
An explanation of central pathways in the model, showing circumstances in which insulin
signaling influences the pathology of AD. The system was modeled using the biochemical
pathway visualization program CellDesigner.
Model 7: Insulin signaling and inflammatory pathways in AD
2.2.1 Insulin Signaling and Inflammation
Insulin signaling in Alzheimer's disease (Model 7) begins when an insulin molecule binds to the
insulin receptor, which activates the insulin receptor substrate and begins a signaling cascade. In
addition to insulin resistance from inflammation, as seen in Parkinson's disease, amyloid beta
oligomers can interfere with insulin signaling by interacting with insulin receptors and
preventing insulin from reaching the receptor (DiCarlo et al., 2010). Microglia and astrocytes
play a major role in the inflammatory process in Alzheimer's disease through the activation of
NF-kB, a transcription factor that promotes the expression of inflammatory cytokines including
IL-6, IL-1β, and TNFα (Hayashi et al., 1993). NF-kB also promotes the expression of the
enzymes iNOS and COX2, which catalyze reactions that produce NO and O2-, respectively
(Arias-Salvatierra et al., 2011; Chandel et al., 2000; Cogswell et al., 1994; Heyen et al., 2000;
Kaltschmidt et al., 2002; Van Wagoner et al., 1999). These reactive molecules also contribute to
inflammation (Glass et al., 2010; Mander et al., 2005).
2.2.2 ROS and Mitochondrial Dysfunction
NO created by iNOS during the inflammatory process (Model 8) is able to diffuse across cell
membranes and react with O2- in the neuron, creating the highly toxic ROS ONOO- (Beckman,
Crow, 1993). The O2- can also degrade into H2O2, and subsequently OH-, both of which are also
harmful, or instead from H2O2 to water in the presence of glutathione (Evans et al., 2002). The
primary source of O2- is the mitochondria, where it is produced in small amounts during normal
respiratory functions. During mitochondrial dysfunction, especially in the respiratory complex I,
larger amounts of O2- are formed (de la Monte et al., 2014). This oxidative stress in turn creates
further dysfunction in complex I, which can then lead to mitochondrial membrane
depolarization. When the mitochondrial membrane is compromised, the H+ ions can leak
through, and the H+ gradient that drives ATP-synthase is weakened, causing ATP depletion.
Without ATP, the cell has no way of transferring energy and will soon die. (Atlante et al., 2008).
Mitochondrial membrane depolarization can also be triggered by the truncation of the
mitochondrial membrane protein BID by the proteins caspase 8 and calpain (Di Carlo et al.,
2010; Kelly et al., 2006; Snyder et al., 2009).
Model 8: Reactive oxygen species synthesis and causes and consequences of mitochondrial
dysfunction in AD
2.2.3 Ca2+ Dysregulation
Ca2+ is an important signaling molecule in neurons, but can be harmful at high concentrations.
Ca2+ usually enters the cytosol from the extracellular environment (Model 9). Ca2+ is also
sequestered in the endoplasmic reticulum. During endoplasmic reticulum stress, which can be
Model 9: Causes and consequences of abnormal Ca2+ signaling in AD cells
triggered by events such as an accumulation of misfolded proteins, the Ca2+ stores can be
released (Casley et al., 2009; Katayama et al., 2004). During Alzheimer's disease conditions, Aβ
oligomers can interact with the Ca2+ voltage-gated ion channels and NMDA receptors to
dysregulate Ca2+ signaling (DeWachter et al., 2009; Mota et al., 2014; Nicholson et al., 2009;
Snyder et al., 2005; Wang et al., 2005). Once Ca2+ levels in the cytosol become too high, the
Ca2+ can activate the protein calpain, which can increase the accumulation of both Aβ oligomers
and neurofibrillary tangles (Goñi-Oliver et al., 2007; Kelly et al., 2006). The Ca2+ can also enter
the mitochondria, which it can activate NADPH oxidase, which promotes the formation of O2
during the oxidation of NADPH to NADP+, contributing to mitochondrial dysfunction
(Stutzmann, 2007).
2.2.4 Neurofibrillary Tangles
Neurofibrillary tangles are comprised primarily of hyperphosphorylated tau proteins, which
usually function to stabilize the cytoskeletal microtubules (Model 10). When the tau are
hyperphosphorylated, however, they release from the microtubules and aggregate into
neurofibrillary tangles. Calpain, p38, and GSK-3β, which is inhibited by the insulin signaling
kinase cascade, can all contribute to tau hyperphosphorylation (Johnson et al., 1989; Li et al.,
2003). Once the tau proteins are phosphorylated, inflammatory conditions create an environment
suitable to aggregation (Liu et al., 2014). Phosphorylated insulin receptor substrate can also
relocate from the nucleus to colocalize with neurofibrillary tangles. Aβ oligomers can activate
Model 10: Neurofibrillary tangle production and triggers in AD
the nuclear protein Jun, which then phosphorylates the insulin receptor substrate (Ma et al.,
2.2.5 β-secretase and Aβ Oligomers
APP expression is promoted by IL-1β signaling from the glia during inflammation (Model 11).
Il-1β also increases the rate of APP cleavage into Aβ fragments, which is catalyzed by the
enzyme β-secretase (Li et al., 2003). β-secretase expression is promoted by NF-kB in neurons
during Alzheimer's disease conditions, as well as by inflammation (Bourne et al., 2007; Heneka
et al., 2005). Once it is created, it can then be activated by calpain after Ca2+ dysregulation
(Goñi-Oliver et al., 2007). Aβ oligomerization is promoted by GSK-3β, which is inhibited by
insulin signaling. Once the Aβ oligomers are created, however, they inhibit further insulin
signaling (Di Carlo et al., 2010). Aβ oligomers have recently been identified as the toxic species
of Aβ in Alzheimer's disease instead of plaques. They contribute to further Ca2+ dysregulation
Model 11: Production of activated beta secretase and consequent formation of amyloid beta
oligomers and onset of their toxic effects in AD
as previously discussed. Lysosome permeability occurs when the lysosomes attempt to degrade
Aβ but become overwhelmed. This releases other toxic lysosomal content into the cytosol
causing further stress to the cell (De Kimpe et al., 2013).
2.2.6 p38 Phosphorylation and Cell Death
The protein p38 plays multiple roles in initiating apoptotic processes in distressed cells. It can be
inhibited by insulin signaling, but activated by inflammation via IL-1β as well as the ROS NO
and O2- (Li et al., 2003). p38 blocks synaptic signaling by preventing the actions of the synaptic
protein synaptophysin. It also acts to phosphorylated tau, contributing to the production of
neurofibrillary tangles. It also converts procaspase 9 to caspase 9, which then joins the
apoptosome and triggers apoptosis, as well as converts procaspase 8 to caspase 8, causing
Model 12: Cellular processes contributing to p38 phosphorylation and neuronal death in AD
mitochondrial stress (Di Carlo et al., 2008). Cell death can be caused by many of the processes
controlled by p38, including ROS from mitochondrial dysfunction, neurofibrillary tangle
buildup, and apoptosis (Liu et al., 2014; Mota et al., 2014). Endoplasmic reticulum stress from
Ca2+ dysregulation and lysosome permeability from Aβ oligomer degradation attempts are also
contributing factors (De Kimpe et al., 2013; Katayama et al., 2004).
2.3 Biochemical Systems Theory
The system was mathematized using BST to create a dynamic model in MATLAB of the species
concentrations and reaction rates as they change over the course of the disease. For further
information on the use of BST to model neurodegenerative disease systems using PLAS, see this
lab's previous work (Broome, Coleman, 2011; Sass et al., 2009; Yeager, Coleman, 2010).
2.3.1 Initial Values
Initial values are assigned to both independent and dependent variables. Independent variables
are species that are not the products of any process or reaction in the system, whereas dependent
variables are the product of at least one process or reaction. The values are assigned on a relative
rather than absolute basis which allows for the estimation of unknown values. When available,
values from the literature are taken into consideration and adapted to the relative scale.
2.3.2 Flux Equations
Flux equations, each of which is assigned a location in the matrix J, describe the relative reaction
rates. Each flux equation takes into account the concentrations of the reactant species and a rate
constant or rate equation. Every reaction or process in the model is assigned a flux equation. Flux
equations take the following form: J(88) = X(258)*Xind(75). In this case, J(88) represents L-
DOPA synthesis, the 88th value in J. Xind(75) represents the independent variable tyrosine, and
X(258) represents the rate ODE. Rate equations consist of a constant multiplied by the sum of all
promoters and inhibitors, where promoters have positive concentration values and inhibitors
have negative concentration values. In this case, X(258) = 0.0001*X(92), where X(92) represents
the promoter phosphorylated tyrosine hydroxylase. In the event that a reaction or process does
not include any promoters or inhibitors, a rate constant, specified as a value in the array k, is
included instead. An example of an unmodified reaction is the equation for the formation of the
IAP inhibition complex: J(96) = k(54)*Xind(82)*X(102), where k(54) is the rate constant,
X(102) is cytosolic SMAC/DIABLO, and Xind(82) is IAP.
2.3.3 Systems Equations
Each dependent variable is assigned a systems equation, which describes the relative
concentration of the species. Systems equations consist of the sum of all the flux equations in
which the dependent variable is a reactant subtracted from the sum of all flux equations in which
the dependent variable is a product. For example, X(93) = X(258)*Xind(75) – X(259)*X(93),
where X(93) represents L-DOPA, X(258)*Xind(75) represents L-DOPA synthesis from tyrosine,
and X(259)*X(93) represents dopamine synthesis from L-DOPA.
2.3.4 Data Analysis
This system exists in three states: a baseline state, a disease state, and a treatment state. In order
to analyze the effects of neurodegeneration on the system, the baseline state is subtracted from
the disease state so that any species with a positive value is elevated during the disease and any
species with a negative value means the species is less present in neurodegeneration. The
treatment state is compared to the baseline state in the same manner. Two treatment states, one in
which the treatment is introduced at disease onset, and one in which treatment is introduced
halfway through disease progression (t = 75), are created. This delay is a novel technique in the
evaluation of treatments in neurodegeneration using BST, and provides significant insight into
the efficacy of the treatment states. The delay is a more realistic simulation of the effects the
treatments will have on the patient, as diagnosis and subsequent treatment at disease onset is
unlikely to occur. The comparative disease state and two comparative treatment states are then
Figure 1: Insulin Signaling in PD A comparison between a Parkinson disease model that
initially includes insulin resistance and one that begins with normal insulin signaling.
graphed together in order to assess the impact of the treatment strategy on the disease both in the
best-case scenario and the more realistic scenario.
3. PD Results
3.1 Insulin Signaling and Inflammation
3.1.1 Disease State
Insulin signaling is impaired by decreasing the rate at which it binds to the insulin receptor. The
relative concentration of the insulin receptor complex continues to decrease as the disease
progresses. Normal insulin signaling also decreases compared to the baseline in response to the
Figure 2: Inflammation in PD An increase in inflammation which is to some extent corrected
by the treatment state.
effects of PD (Fig.1). Inflammation increases in the PD state compared to the baseline state as a
result of an increase in ROS, NF- κB signaling in the astrocytes and microglia, and cytokine
expression (Fig. 2).
3.1.2 Treatment State
The lack of insulin signaling is partially corrected for by the inclusion of pioglitazone and
sodium butyrate, which act as insulin sensitizers (Gao et al., 2009; Hu et al., 2013). Insulin
signaling is fully recovered in the model when it is initially at normal levels, and is significantly
recovered in the insulin resistant model as well during full treatment (Fig. 1). With a delayed
initiation of treatment, insulin signaling in both the initially insulin resistant and initially insulin
sensitive models are partially corrected but continue on a downward trend. Treatments of vitamin
D (Chang et al., 2004), NAC, and aspirin (Ambhore et al., 2014) correct inflammation. Vitamin
D inhibits NOS from producing pro-inflammatory NO in the glia. NAC inhibits NF- κB from
entering the nucleus (Oka et al., 2000), thereby reducing expression of inflammatory cytokines.
NAC also acts as a COX2 inhibitor, reducing the production of prostaglandins. Aspirin
acetylates COX2, inhibiting its function as a promoter of prostaglandin synthesis. The decrease
in cytokine, NO, and prostaglandin concentrations accordingly decrease inflammation, although
inflammation is still largely present (Fig. 2). The full treatment state was similar to the delayed
state in the prevention of inflammation.
Figure 3: Cytosolic Dopamine in PD Increased levels of cytosolic dopamine levels which are
eventually fully rectified in the treatment state.
3.2 Dopamine
3.2.1 Disease State
Dopamine levels are manipulated in the disease state by decreasing the concentration of VMAT
transporters that allow the dopamine to be sequestered into vesicles. Extracellular dopamine
concentrations are reduced to account for the degeneration of nearby dopaminergic neurons from
PD, thereby reducing intercellular dopamine signaling. Dopamine degradation rates are increased
in the disease state. Cytosolic dopamine levels actually increase in the PD model (Fig. 3);
however, vesicle dopamine is decreased (Fig. 4). Cytosolic dopamine is undesirable in the cell
because it is vulnerable to degradation into ROS. Vesicle dopamine is the preferred form because
it is protected from degradation and is available for signaling at the synapse.
Figure 4: Vesicle Dopamine in PD The decrease in cytosolic dopamine levels in the disease
state is prevented in the treatment state, and the vesicle concentration becomes more constant.
3.2.2 Treatment State
Treatments for dopamine imbalances in the PD neuron include vitamin D (Puchacz et al., 1996)
and edaravone (Xiong et al., 2011). These two treatments complement each other because
vitamin D increases cytosolic dopamine levels by increasing expression of tyrosine hydroxylase,
which is important in dopamine synthesis, while edaravone promotes VMAT expression,
increasing the ability of dopamine to be transported to the vesicles. The treatment state initially
follows the disease state progression with a sharp increase in cytosolic dopamine levels and
corresponding decrease in dopamine vesicle concentration, but then corrects to normal cytosolic
dopamine levels (Fig. 3). Full treatment returns to normal levels much more quickly than delayed
treatment does, and delayed treatment reaches the same maximum concentration difference as
the disease state before declining. Only a slight decrease from baseline in vesicle dopamine
concentration is observed with full treatment (Fig. 4). Delayed treatment allows for a much
larger gap from the baseline that is never recovered within the scope of the model.
Figure 5: Reactive Oxygen Species in PD The level of reactive oxygen species is much lower
in the treatment state than the disease state.
3.3 ROS and RNS
3.3.1 Disease State
Mitochondrial dysfunction, dopamine metabolism rate, ROS production rates, and RNS
production rates are increased in the disease state while antioxidant concentrations are decreased
to trigger ROS and RNS accumulation. The ROS and RNS portrayed in Figure 5 consist of the
sums of the levels of cytosolic O2-, H2O2, OH-, ONOO-, NO, and mitochondrial O2- in the
neuron, as well as extracellular NO.
3.3.2 Treatment State
Antioxidant treatments included in the model are vitamin D, edaravone, and NAC (Samuni et al.,
2013). Vitamin D can help to decrease cytosolic O2- concentrations, and prevent Ca2+ influx into
the cell. Ca2+ buildup in the mitochondria can cause excess O2- production (Gandhi et al., 2009).
Edaravone prevents mitochondrial DNA damage, reducing mitochondrial dysfunction (Xiong et
al., 2011). NAC acts as an antioxidant in several ways. It is a precursor to glutathione, which is
important in neutralizing H2O2. It can also react with ONOO- to degrade it into NO2- and OH-,
and then react with those two species to form HNO2 and water. Full treatment greatly reduced
ROS and RNS presence in the neuron (Fig. 5). Delayed treatment originally follows the course
of the disease state, but then begins to approach the full treatment state.
3.4 Tau Phosphorylation, Neurofibrillary Tangles, and Lewy Bodies
3.4.1 Disease State
Tau phosphorylation is triggered by increasing activated GSK-3β concentrations by decreasing
insulin signaling, indirectly triggering p38 phosphorylation, and increasing α-synuclein
aggregations, which interact with tau and create toxic accumulations of the two proteins (Lei et
al., 2010). Hyperphosphorylated tau in the disease state increases sharply compared to the
baseline state (Fig. 6). Neurofibrillary tangles are a consequence of tau hyperphosphorylation
and increase accordingly (Fig. 7). Lewy bodies are the result of aggregated α-synuclein,
hyperphosphorylated tau, and other misfolded proteins, and increase in concentration as these
species become more numerous (Fig. 8).
Figure 6: Phosphorylated Tau in PD Tau Phosphorylation is elevated in Parkinson's disease,
but is slowed in the treatment state.
3.4.2 Treatment State
Treatments included in the model that affect tau phosphorylation, neurofibrillary tangles, and
Lewy bodies are lithium (Petit-Paitel et al., 2009), edaravone (Zhou et al., 2013), and sodium
butyrate (Taylor et al., 2013). Lithium promotes GSK-3β phosphorylation, which prevents it
from phosphorylating tau. Edaravone also inhibits tau phosphorylation. Sodium butyrate triggers
the expression of MKP-1, which prevents tau phosphorylation by inhibiting p38 activation.
Phosphorylated tau is considerably diminished in the treatment state, but is still present (Fig. 6).
Neurofibrillary tangles (Fig. 7) and Lewy bodies (Fig. 8) decline at a greater rate. In these three
cases, a delayed treatment does cause a decrease in species concentration, but still continues the
Figure 7: Neurofibrillary Tangles in PD Neurofibrillary tangles are increasingly present in
the disease state, but are present at a lower level in the treatment state.
upward trend characteristic of the disease state. Delayed treatment slows the accumulation of
these species but does not prevent it.
Figure 8: Lewy Bodies in PD Lewy body increase in Parkinson's disease is lessened in the
treatment state.
3.5 p38 Phosphorylation
3.5.1 Disease State
Decreasing the rate of insulin signaling, increasing the concentrations of ROS, and increasing
the presence of prostaglandin J2 from the baseline state increases the activation of p38 in the
disease state (Fig. 9). A decrease in the availability of parkin as an E3 ubiquitin ligase in the p38
degradation pathway is also a contributing factor to the excess p38 phosphorylation. As a result
of p38 phosphorylation, mitochondrial dysfunction, apoptosis, tau phosphorylation, dopamine
synthesis, and Hsp27 activation also increase.
Figure 9: Phosphorylated p38 in PD The disease state includes increased phosphorylation of
p38, which is attenuated in the treatment state.
3.5.2 Treatment State
The full treatment state reduces p38 concentrations past the baseline state (Fig. 9). Activation of
MKP-1 through sodium butyrate treatment inhibits p38 phosphorylation (Taylor et al., 2013).
Sodium butyrate increases expression of the DUSP1 gene, increasing the expression of MKP-1.
This phosphatase is responsible for dephosphorylating p38, rendering it inactive. The reduction
in ROS levels also contributed to the decline of the p38 concentration, as they are less available
to promote the phosphorylation. Delayed treatment failed to produce similar results. Instead, it
predicted an increase in phosphorylated p38 compared to the baseline state, but still produced a
positive impact by remaining at a lower concentration than in the disease state.
3.6 Cell Death
3.6.1 Disease State
Cell death can be triggered by apoptosome formation following activation of the caspase cascade
by ROS in the cytosol, or by mitochondrial dysfunction and release of cytochrome c. Both
cytosolic and mitochondrial ROS are elevated in the disease state. Lewy bodies and
Figure 10: Cell Death in PD The treatment state corrects for some of the cell death caused by
pathways influenced by insulin signaling in Parkinson's disease.
neurofibrillary tangles can also cause cell death. Finally, DNA fragmentation and chromosome
condensation caused by AIF presence in the nucleus can lead to apoptosis. AIF is released along
with cytochrome c as the PTP opens on the mitochondrial membrane. These factors all
contribute to the increased probability of apoptosis seen as the model progresses (Fig.10).
3.6.2 Treatment State
Apoptosis is prevented by several drugs including edaravone (Cheng et al., 2014; Xiong et al.,
2011), Sodium butyrate (Taylor et al., 2013), cyclosporin A, and nortriptyline (Lamarche et al.,
2012). Edaravone prevents apoptosis by inhibiting tau phosphorylation, promoting the
expression of the mitochondrial protein Bcl-XL, and inhibiting the expression of the related Bax
protein. The Bax protein binds to the truncated version of Bid and increases opening of the PTP.
Bcl-XL can bind to the Bax/Bid complex to prevent this process. Sodium butyrate inhibits the
apoptotic proteins Jun and p38 from being activated through its promotion of MKP-1 expression.
Cyclosporin A and nortriptyline prevent the PTP from opening and releasing apoptotic factors
such as cytochrome c, SMAC/DIABLO, and AIF. Cell death is successfully prevented if the
treatment is presented at disease onset, but is still present, albeit in a diminished state, when the
treatment is delayed.
4. AD Results
4.1 Insulin Signaling and Inflammation
4.1.1 Disease State
Insulin resistance (Fig. 11) was achieved by decreasing the rate at which insulin bound to its
receptor and at which the receptor substrate was activated. The condition was further exacerbated
in the disease state with the onset of inflammation and the presence of Aβ oligomers, which
compete with insulin for receptor interactions. Inflammation (Fig. 12) is triggered in the glia by
the presence of ROS and the release of NF-kB from its inhibitor, allowing it to enter the nucleus
and begin its actions as a transcription factor for pro-inflammatory cytokines and enzymes that
produce even more ROS.
4.1.2 Treatment State
Figure 11: Insulin Signaling in AD Progressive insulin signaling accompanying AD is
attenuated with insulin sensitizing treatment
Insulin sensitization (Fig. 11) is achieved in this model by the inclusion of pioglitazone and
sodium butyrate, which increase interactions between insulin and its receptor (Gao et al., 2009;
Heneka et al., 2005). Pioglitazone and sodium butyrate are successful in recovering normal
insulin signaling in both a full treatment scenario and recover almost all signaling in the delayed
treatment model, although because the signaling had already been further decreased, it acted
more slowly than the full treatment. This model does not include a treatment for inflammation
directly, and once NF-kB has been activated by ROS, the presence of antioxidants, which the
model does include, will not necessarily stop inflammation triggered by the newly expressed pro-
Figure 12: Inflammation in AD Inflammatory processes in AD increase as the disease
progresses
inflammatory cytokines. The inflammation does decrease slightly from the reduction of ROS
4.2 ROS and Mitochondrial Dysfunction
4.2.1 Disease State
The concentration of ROS in the model was determined by summing the concentrations of all
ROS in the neuron, astrocyte, and microglia and include O2-, H2O2, OH-, NO, and ONOO-. In
the disease model, the ROS steadily increase during the progression of the disease state (Fig. 13).
ROS contribute to cell damage by reacting with lipids, proteins, and DNA and altering or
Figure 13: Reactive Oxygen Species in AD O -, H O , OH-, and ONOO- in neurons,
astrocytes, and microglia all increase with AD
preventing their function. Mitochondria are especially vulnerable to ROS toxicity because
respiration is a major source of ROS in the cell. Mitochondrial dysfunction data shown (Fig. 14)
includes both complex I dysfunction and mitochondrial membrane depolarization, which both
interfere with normal respiratory function and contribute to further production of ROS. In the
model, mitochondrial dysfunction increases at an increasing rate over the course of AD.
4.2.2 Treatment State
The treatment strategy for mitochondrial dysfunction focused on preventing ROS buildup. The
antioxidant curcumin was chosen for this model. Curcumin is a naturally occurring substance
Figure 14: Mitochondrial Dysfunction in AD Complex I dysfunction and mitochondrial
membrane depolarization can both be prevented by the neutralization of reactive oxygen
species
that is present in the spice turmeric, and is responsible for its characteristic yellow color.
Curcumin is able to neutralize two ROS per molecule by donating H+ to the ROS, rendering
them less reactive (Barzegar 2012). The curcumin is able to reduce the amounts of ROS in the
system, but not completely erase their presence (Fig. 13). Interestingly, delayed treatment is able
to quickly reduce ROS concentrations to full treatment levels. This does not, however, transfer to
mitochondrial dysfunction (Fig. 14). Almost all mitochondrial dysfunction is able to be
attenuated by the presence of an antioxidant if it is administered at the onset of the functional
Figure 15: Ca2+ Dysregulation in AD Ca increases in toxic amounts in Alzheimer's disease,
which can be prevented by inhibition of Ca entering the cell from the extracellular environment
deterioration. If it is administered after some dysfunction has already occurred, however, it can
delay but not prevent further respiratory inhibition.
4.3 Ca2+ Dysregulation
4.3.1 Disease State
Ca2+ influx can occur from two sources in the model: entry into the cell from ion channels and
release of Ca2+ stores in the endoplasmic reticulum in response to stressors. Both of these
processes are activated in AD with the result that the intracellular Ca2+ concentration increases
during the disease state (Fig. 15), leading to abnormal Ca2+ signaling that contributes further to
disease progression.
4.3.1 Treatment State
The treatment state focused on preventing Ca2+ influx from ion channels, not preventing
endoplasmic reticulum Ca2+ release. The channel blocker MRS2481 is included in the model to
this effect (Diaz et al., 2009). If added before any abnormal Ca2+ signaling, the inhibitor
effectively prevents an overabundance of Ca2+ in the cytosol (Fig. 15). If Ca2+ has already started
to accumulate, however, the treatment can only partially block further dysregulation. This
additional Ca2+ may derive from the endoplasmic reticulum, as too much cytosolic Ca2+ can
cause the endoplasmic reticulum to release its Ca2+ stores, further exacerbating the aberrant
signaling (Casley et al., 2009).
Figure 16: Neurofibrillary Tangles in AD Neurofibrillary tangles increase during AD, but can
be prevented primarily by inhibiting tau phosphorylation
4.4 Neurofibrillary Tangles
4.4.1 Disease State
Neurofibrillary tangles are increased in the Ad model compared to the baseline in several ways.
The increased insulin resistance prevents a kinase cascade that blocks GSK-3β, which can
phosphorylate tau. Additionally, Ca2+ increases activate calpain to further increase tau
hyperphosphorylation. Finally, ROS can cause inflammation and p38 activation, both of which
can contribute to tau hyperphosphorylation. These hyperphosphorylated tau are then able to
accumulate in neurofibrillary tangles, which are present in increasing amounts in the AD model
4.4.2 Treatment State
The treatment state includes several methods of reducing neurofibrillary tangle production.
Curcumin as an antioxidant prevents some inflammation and p38 phosphorylation. Pioglitazone
can inhibit GSK-3β activity by increasing insulin signaling. MRS2481 reduces calpain activation
by reducing Ca2+ signaling. With the final addition of a direct GSK-3β inhibitor, lithium chloride
(Noble et al., 2005), neurofibrillary tangles are dramatically decreased in the full treatment
Figure 17: Beta Secretase in AD Increases in beta secretase concentrations are corrected in the
treatment state
model (Fig. 16). In the delayed treatment state, the neurofibrillary tangle formation process can
only be partially corrected.
4.5 β-secretase and Aβ Oligomers
4.5.1 Disease State
β-secretase is increased in the model (Fig. 17) through the increase in inflammation and
indirectly by Ca2+ signaling from an increase in calpain. Accordingly, Aβ oligomers are
increased (Fig. 18) through the increased production of Aβ from β-secretase increase, amplified
Figure 18: Amyloid Beta Oligomers in AD Amyloid beta oligomers are greatly reduced in the
treatment state compared to the disease state
APP expression from glial IL-1β signaling as a result of inflammatory signaling, and greater
GSK-3β activity from a lack of insulin signaling.
4.5.2 Treatment State
β-secretase is treated in the model through pioglitazone, which acts as a β–secretase inhibitor as
well as an insulin sensitizer (Heneka et al., 2005). This treatment decreases β–secretase to near
normal levels after an initial rise in concentration in the full treatment state (Fig. 17), and
provides some inhibitory behavior, although to a lesser extent, in the delayed treatment model.
Aβ oligomers sharply decrease in the full treatment state (Fig. 18). This is a result of a lack of Aβ
Figure 19: p38 Phosphorylation in AD p38 is increasingly phosphorylated as AD progresses,
but can be inhibited in the treatment state
availability because of β-secretase scarcity, as well as inflammation decreases from curcumin's
antioxidant properties and recovered insulin signaling from pioglitazone sensitization. Curcumin
also more directly inhibits Aβ aggregation (Ganugapati et al., 2015) by interacting with the
active site of the APP. In the delayed treatment state, the oligomers also decrease below disease
levels, but not to the extent of the full treatment.
4.6 Phosphorylated p38 and Cell Death
4.6.1 Disease State
Figure 20: Cell Death in AD Cell death increases throughout disease progression, but is
somewhat attenuated in the treatment state.
p38 increases decidedly as the disease state advances (Fig. 19). This is a result of an increased
number of ROS and inflammation accompanied by a decrease in insulin signaling. p83 activation
triggers cell death in several ways, contributing to the increase in degeneration in the disease
state (Fig. 20). Endoplasmic reticulum stress from abnormal cytosolic Ca2+ accumulation,
lysosome permeability, and ROS more directly also contribute to the increasing likelihood that a
neuron will die.
4.6.2 Treatment State
While p38 activation initially increases even during the full treatment state, it begins decreasing
about halfway through the model (Fig. 19). The delayed treatment state also includes a decrease
in p38 phosphorylation, but to a lesser extent. Curcumin as an antioxidant and pioglitazone as an
insulin sensitizer help to prevent p38 activation, and an additional p38 inhibitor, sodium butyrate,
is also included in the model (Taylor et al., 2013). Cell death is affected by every process
previously discussed, and therefore every treatment included in the model works to ultimately
prevent degeneration. This effort is somewhat successful, as the full treatment state decreases
cell death, and improves slightly in efficacy as the model progresses (Fig. 20). The delayed
treatment, however, is much less effective.
5. Discussion
5.1.1 Disease State
The disease state model mimics the current understanding of PD in that it shows increases in
inflammation, apoptotic factors, ROS and RNS, abnormal dopamine concentrations, and elevated
levels of phosphorylated tau, neurofibrillary tangles, and Lewy bodies, which are characteristic
of PD. These factors are only a fraction of the numerous of processes that typify PD, which also
include but are not limited to the misfolding of α-synuclein, gene mutations, endoplasmic
reticulum stress, and ubiquitin proteasome system malfunction, which are not fully explored in
this model. The disease state modeled here is exacerbated by a loss of insulin signaling, usually
caused by an excess of fats and sugars in the diet and a major factor in the development of
T2DM.The model also suggests that dopaminergic neuronal insulin signaling is highly impaired
in PD. This is likely caused by PD effects such as chronic inflammation and oxidative stress,
which are common causes of insulin resistance in T2DM patients (Evans et al., 2002).
5.1.2 Treatment State
The model suggests a variety of treatment options to ameliorate both causes and symptoms of
PD. Vitamin D, Li, aspirin, NAC, edaravone, pioglitazone, Sodium butyrate, cyclosporin A, and
nortriptyline are the treatments included in the version of the model presented in this paper.
Vitamin D acts as a ROS scavenger as well as decreasing inflammation. Lithium is a GSK 3β
inhibitor. It is currently used as a treatment for bipolar disorder, but is being researched as a
treatment for PD. Aspirin, a common, over-the-counter pain medication, is a NSAID and inhibits
the inflammatory cytokine COX2. NAC acts as both an anti-inflammatory and antioxidant
compound by inhibiting NF-κB, increasing glutathione concentrations, and reacting to neutralize
ROS and RNS (Model 13). NAC is currently used as a treatment for a variety of psychiatric
conditions including schizophrenia, obsessive-compulsive disorder, and addiction (Dean et al.,
2011). Edaravone protects against mitochondrial damage and tau phosphorylation, while
promoting dopamine entry into vesicles. It is currently being investigated as a PD medication, as
well as being involved in ongoing clinical trials to evaluate its potential as a treatment for ALS,
another neurodegenerative disease. Pioglitazone is a known insulin sensitizer and is used in the
treatment of T2DM. Sodium butyrate prevents apoptosis by inhibiting the activation of pro-
apoptotic proteins p38 and Jun. It also acts as an insulin sensitizer, and is currently being studied
as a treatment for T2DM and the neurodegenerative condition Huntington's disease. Cyclosporin
A, an immunosuppressant, and nortriptyline, an antidepressant, both block apoptosis by
preventing the PTP from opening.
PD symptoms occur only after the disease progression is well under way. Treatment, therefore, is
often delayed. This model includes one condition in which treatment is introduced at the onset of
the disease, and one in which treatment is introduced halfway through the modeled disease
progression. The differences between the scenarios highlight the importance of early detection of
PD and, for the familial version of the disease, the introduction of possible preventative
measures. The delayed treatment states for insulin resistance (Fig. 1), cell death (Fig. 10), p38
phosphorylation (Fig. 9), Lewy bodies (Fig. 8), neurofibrillary tangles (Fig. 7), and tau
Model 13: Example of treatment action—NAC as an antioxidant and NF-κB inhibitor
in PD
phosphorylation (Fig. 6) each resemble their disease state simulations more than their full
treatment states. ROS (Fig. 5) and inflammation (Fig. 2) levels are partially corrected, but still
high. Cytosolic dopamine (Fig. 3) levels eventually return to normal, and while vesicle dopamine
(Fig. 4) levels are stabilized with treatment, the original concentration was not restored.
5.2.1 Disease State
The disease state accurately accounts for many of the pathological trends observed in AD brains,
including increases in inflammation, cytosolic Ca2+, ROS and mitochondrial dysfunction,
neurofibrillary tangles, Aβ oligomers, and cell death. Insulin resistance is connected to all of
these changes and is a well characterized aspect of AD. Insulin signaling, or a lack thereof,
affects the two main downstream components of AD, neurofibrillary tangles and Aβ oligomers,
both of which are composed of pathologically modified proteins or peptide fragments. Both
T2DM and T1DM effects, insulin receptor desensitization and a lack of insulin, are observed in
AD conditions, further linking neurodegeneration to metabolic defects.
5.2.2 Treatment State
The approach to the AD treatment state was somewhat different than in the PD model. The PD
model was more concerned with suggesting pathways to treat rather than specific compounds to
treat them, although specific compounds were suggested. The AD model suggests five treatments
instead of ten, representing a more realistic, although still complicated, drug cocktail. The AD
treatments were chosen in many cases on their ability to affect multiple implicated pathways,
allowing for a reduction in total treatments included. This approach minimizes the likelihood of
severe side effects and drug interactions. These aspects were not investigated in the model,
however, because the focus is on the pathways themselves, not specific treatments. If two of the
treatments suggested happen to interact with each other, for instance, other compounds can be
found that inhibit the same processes.
This model included several treatments for AD that overlap with PD treatment suggestions.
Sodium butyrate is able to sensitize the insulin receptor as well as prevent p38 activation, which
are important processes that require intervention in both models. Lithium also acts more
downstream of the insulin receptor to reduce tau hyperphosphorylation in both models.
Pioglitazone was incorporated in the PD model as an insulin sensitizer only, but also acts as a β-
secretase inhibitor in the AD model. The molecule MRS2481 is a Ca2+ channel blocker and
prevents an influx of Ca2+. Curcumin is a potent naturally occurring antioxidant molecule that is
currently being investigated as a treatment for AD. Not only does it neutralize ROS, but it also
prevents Aβ oligomerization by binding to the Aβ active site. Taken together, these treatments
all contribute to the rectification of the multiple biochemical pathways affected by insulin
resistance in AD.
5.3 MATLAB
This paper introduces a new protocol for coding neurodegenerative systems using MATLAB
instead of PLAS. Unlike PLAS, MATLAB is capable of analyzing all aspects of the quantitative
model, from solving the ODEs to preparing graphs. PLAS requires the use of separate programs
such as Microsoft Excel for any data analysis and visualization required after the ODEs are
solved. MATLAB is also able to take into account multiple qualitative outcome possibilities for
a single species. In other words, MATLAB is able to recognize a condition, such as a species
reaching a threshold concentration, and substitute the original equation with an alternate one
leading to a qualitatively different outcome once the species reaches the threshold. Another
advantage of MATLAB is the ability to order the solutions to the equations so that the species
most sensitive to the change are immediately apparent. Finally, MATLAB can introduce new
events, such as introducing or altering treatment dosages, at any time during the simulation.
PLAS is only able to use the initial values given at the onset of the simulation. Overall,
MATLAB is the more flexible and efficient analytical tool.
6. Conclusion
The two models described in this paper explore the reciprocal intensification of PD and insulin
resistance and AD and insulin resistance through a mathematical lens. They provide a quick,
cost-effective means of evaluating possible neurodegenerative disease treatment options for
patients who also exhibit insulin resistance, and suggests mechanisms for the interactions
between the conditions. The PD model emphasizes dopaminergic neuron deterioration through
ROS production via dysfunctional mitochondria and dopamine metabolism as well as Lewy body
and neurofibrillary tangle production through hyperphosphorylated tau accumulation. The model
also simulates the reduction in insulin signaling that occurs as a result of the chronic
inflammation and ROS production that can accompany PD. Insulin signaling is partially lost
even when it is not initially impaired. The AD model focuses on insulin signaling impairment
through inflammatory processes, including ROS production and accompanying mitochondrial
production, as well as Ca2+ dysregulation, the characteristic AD pathological components
neurofibrillary tangles and Aβ oligomers, and lastly p38 phosphorylation and cell death.
Finally, the models evaluates the neuroprotective ability of a variety of treatments, both
experimental and currently available to treat PD, AD, or T2DM. The models predict that a
combined approach, initiated as early as possible and targeting a wide range of pathways, is the
Acknowledgements:
This work was supported by grants from the Howard Hughes Medical Institute, Undergraduate
Biological Sciences Education Program, The College of William and Mary; and by The Roy R.
Charles Center for Academic Excellence at The College of William and Mary, by Douglas
Morton and Marilyn Brown.
I would also like to thank Josh Burk and Debbie Noonan for agreeing to be a part of my thesis
committee, as well as Randy Coleman for enthusiastically supporting this work from the very
beginning.
The qualitative models were developed using the program CellDesigner version 4.3, and can be
found a
The mathematical models were developed using MATLAB R2014a, which could be found at the
following website upon beginning this wor
Abbreviations:
BST, Biochemical Systems Theory; PLAS, Power Law Analysis and Simulation; PD,
Parkinson's Disease; T2DM, Type-II Diabetes Mellitus; T1DM, Type-I Diabetes Mellitus; CNS,
Central Nervous System; ROS, Reactive Oxygen Species; RNS, Reactive Nitrogen Species; AD,
Alzheimer's disease; Aβ, Amyloid Beta; APP, Amyloid Precursor Protein; ODE, Ordinary
Differential Equation; PPARG, Peroxisome-Proliferator-Activated Receptor γ; IL-6, Interleukin-
6; IL-1β, Interleukin-1β; TNFα, Tumor Necrosis Factor α; MCSF, Macrophage Colony-
Stimulating Factor; COX2, Cyclooxygenase 2; NO, Nitric Oxide; H2O2, Hydrogen Peroxide; O2-
, Superoxide Radical; OH-, Hydroxyl Radical; ONOO-, Peroxynitrite; HNO2, Nitrous Acid; NF-
κB, Nuclear Factor κB; VMAT, Vesicular Monoamine Transporter Proteins; SOD2, Superoxide
Dismutase 2; PI3K, Phosphatidylinositol 3 Kinase; PIP2, Phosphatidylinositol Bisphosphate;
PIP3, Phosphatidylinositol Trisphosphate; GSK-3β, Glycogen Synthase Kinase 3β; MKP-1,
Mitogen-Activated Protein Kinase Phosphatase 1; DUPS1, Dual Specificity Protein Phosphatase
1; PTP, Permeability Transition Pore; AIF, Apoptosis-Inducing Factor; SMAC/DIABLO,
Second Mitochondria-Derived Activator of Caspases/Direct IAP Binding Protein with Low pI;
IAP, Inhibitor of Apoptosis; NAC, N-acetylcysteine; NOS, Nitric Oxide Synthase; NSAID, Non-
Steroidal Anti-inflammatory Drug; ALS, Amyotrophic Lateral Sclerosis
References:
Ambhore, N.S., Prasanna, M., Antony, A.S., Kumar, M.N.S., Elango, K. Pharmacological and
anti-oxidant evaluation of Aspirin, nimodipine and its combination for anti-Parkinson's activity
in MPTP induced rat model. Int J Health Allied Sci 2014; 3(1):14-22.
Arias-Salvatierra, D., Silbergeld, E.K., Acosta-Saavedra, L.C., Calderon-Aranda, E.S. Role of nitric oxide produced by iNOS through NF-kB pathway in migration of cerebellar granule neurons induced by Lipopolysaccharide. Cell Signal 2011; 23(2), 425-35.
Arima, K., Hirai, S., Sunohara, N., Aoto, K., Izumiyama, Y., Ueda, K, et al. Cellular co-localization of phosphorylated tau- and NACP/alpha-synuclein-epitopes in lewy bodies in sporadic Parkinson's disease and in dementia with Lewy bodies. Brain Res 1999; 843(1-2):53-61.
Atlante, A., Amadoro, G., Bobba, A., de Bari, A., Corsetti, V., Pappalardo, G., Marra, E., Calissano, P., Passarella, S. A peptide containing residues 26-44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim et Biophys Acta 2008; 1777: 1289-1300.
Bancher, C., Braak, H., Fischer, P., Jellinger, K.A. Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer's and Parkinson's disease patients. Neurosci Lett 1993; 162: 179-82.
Barzegar, A. The role of electron-transfer and H-atom donation on the superb antioxidant activity and free radical reaction of curcumin. Food Chem 2012; 135, 1369-1376.
Beckman, J.S., Crow, J.P. Pathological implications of nitric oxide, superoxide, and peroxynitrite formation. Biochem Soc Trans 1993; 21: 330-4.
Bosco, D., Plastino, M., Cristiano, D., Colica, C., Ermio, C., De Bartolo, M., et al. Dementia is associated with Insulin Resistance in patients with Parkinson's disease. J Neurol Sci 2012; 315(1-2): 39-43.
Broome, T.M., Coleman, R.A. A mathematical model of cell death in multiple sclerosis. J Neurosci Methods 2011; 201(2): 420-5.
Bourne, K.Z., Ferrari, D.C., Lange-Dohna, C., Robner, S., Wood, T.G., Perez-Polo, J.R. Differential Regulation of BACE1 Promoter Activity by Nuclear Factor-kB in Neurons and Glia Upon Exposure to β-Amyloid Peptides. J Neurosci Res. 2007; 85, 1194-1204. Casley, C. S., Lakics, V., Lee, H. G., Broad, L. M., Day, T. A., Cluett, T.,Smith, M. A., O'Neill, M. J., and Kingston, A. E. Up-regulation of astrocyte metabotropic glutamate receptor 5 by amyloid-β peptide. Brain Res. 2009; 1260, 65–67. Chandel, N.S., Trzyna, W.C., McClintock, D.S., Schumacker, P.T. Role of oxidants in NF-kappa B activation and TNF-alpha gene trascription induced by hypoxia and endotoxin. J immunol 2000; 165(2), 1013-21.
Chang, J.M., Kuo, M.C., Kuo, H.T., Hwang, S.J., Tsai, J.C., Chen, et al. 1-α,25-Dihydroxyvitamin D3 regulates inducible nitric oxide synthase messenger RNA expression and nitric oxide release in macrophage-like RAW 264.7 cells. J Lab Clin Med 2004; 143(1): 14-22.
Cheng, B., Guo, Y., Li, C., Ji, B., Pan, Y., Chen, J., Bai, B. Edaravone protected PC12 cells against MPP(+)-cytotoxicity via inhibiting oxidative stress and up-regulating heme oxygenase-1 expression. J Neurol Sci 2014.
Choi, J-S., Oh, J-I., Na, M., Lee, S-K., Joo, S.H. PKCδ promotes etoposide-induced cell death by phosphorylating Hsp27 in HeLa cells. Biochem and Biophys Res Commun 2012; 426 (4): 590-5.
Choi, W-S., Eom, D-S., Han, B.S., Kim, W.K., Han, B.H., Choi, E-J., et al. Phosphorylation of p38 MAPK Induced by Oxidative Stress Is Linked to Activation of Both Caspase-8 and -9-mediated Apoptotic Pathways in Dopaminergic Neurons. J Biol Chem 2004; 279 (19): 20451-60.
Cogswell, J.P., Godlevski, M.M., Wisely, G.B., Clay, W.C., Leesnitzer, L.M., Ways, J.P., … NF-kB Regulates IL-1β Transcription Through a Consensus NF-kB Binding Site and a Nonconsensus CRE-Like Site. J. Immunol 1994; 153(2), 712-23.
Dean, O., Giorlando, F., Berk, M. N-acetylcysteine in psychiatry: current therapeutic evidence and potential mechanisms of action. J Psychiatry Neurosci 2011; 36(2): 78-86.
De Kimpe, L., van Haastert, E.S., Kaminari, A., Zwart, R., Rutjes, H., Hoozemans, J.J.M. Intracellular accumulation of aggregated polyglutamate amyloid beta: convergence of aging and Aβ pathology at the lysosome. AGE 2013; 35, 673-87.
De la Monte, S., Tong, M. Brain metabolic dysfunction at the core of Alzheimer's disease. Biochem Pharm 2014; 88, 548-559.
Dewachter, I., Filipkowski, R. K., Priller, C., Ris, L., Neyton, J., Croes, S.,Terwel, D., Gysemans, M., Devijver, H., Borghgraef, P.,Godaux, E., Kaczmarek,L., Herms, J., and Van Leuven, F. Deregulation of NMDA-receptor function and down-stream signaling in APP [V717I] transgenic mice. Neurobiol. Aging 2009; 30, 241–256. Diaz, J.C., Simakova, O., Jacobson, K.A., Arispe, N., Pollard, H.B. Small molecule blockers of the Alzheimer's Aβ calcium channel potently protect neurons from Aβ toxicity. PNAS 2009; 106(9), 3348-3353.
Di Carlo, M., Picone, P., Carrotta, R., Giacomazza, D., San Biagio, P.L. Insulin Promotes Survival of Amyloid-Beta Oligomers Neuroblastoma Damaged Cells via Caspase 9 Inhibition and Hsp70 Upregulation. J Biomed Biotechnol. 2010; art. #147835.
Evans, J.L., Goldfine, I.D., Maddux, B.A., Grodsky, G.M. Oxidative Stress and Stress-Actived Signaling Pathways: A Unifying Hypothesis of Type 2 Diabetes. Endocr Rev 2002; 23 (5): 599-622.
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., et al. PINK1-Associated Parkinson's Disease Is Caused by Neuronal Vulnerability to Calcium-Induced Cell Death. Mol Cell 2009; 33 (5): 627-38.
Ganugapati, J., Babu, R., Ahuja, S.J., Mukundan, M., Vutukuru, S.S. Scrrening and Molecular Docking Studies of Curcumin and Its Derivatives as Inhibhitors of Amyloid-β Protein: A Key Protein in Alzheimer's Disease. Asian J Pharm Clin Res, 2015; 8(2), 98-101.
Gao, Z., Yin, J., Zhang, J., Ward, R.E., Martin, R.J., Lefevre, M., et al. Butyrate Improves Insulin Sensitivity and Increases Energy Expenditure in Mice. Diabetes 2009; 58(7): 1509-17.
Ghosh, A., Roy, A., Liu, X., Kordower, J.H., Mufson, E.J., Hartley, D.M., et al. Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson's disease. Proc Nat Acad Sci 2007; 104 (47): 18754-9.
Glass, C.K., Saijo, K., Winner, B., Marchetto, M.C., Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010; 140 (6), 918-934
Goñi-Oliver, P., Lucas, J.J., Avila, J., Hernández, F., N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. J Biol Chem 2007; 282(31) 22406–22413. Hayashi, T., Ueno, Y., Okamoto, T. Oxidoreductive Regulation of Nuclear Factor k B: Involvement of a Cellular Reducing Catalyst Thioredoxin. J. Biol. Chem 1993; 268(15), 11380-8.
Heidenreich, K.A., Kummer, J.L. Inhibition of p38 Mitogen-activated Protein Kinase by Insulin in Cultured Fetal Neurons. J Biol Chem 1996; 271 (17): 9891-4.
Heneka, M.T., Sastre, M., Dumitrescu-Ozimek, L., Hanke, A., Dewachter, I., Kuiperi, C., . Acute Treatment with the PPARg agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ 1-42 levels in APPV717I transgenic mice. Brain 2005; 128, 1442-53.
Heyen, J.R.R., Ye, S.M., Finck, B.N., Johnson, R.W. Interleukin (IL)-10 inhibits IL-6 production in microglia by preventing activation of NF-kB. Mol. Brain. Res 2000; 77(1), 138-47.
Hunot, S., Boissiere, F., Faucheux, B., Brugg, B., Mouatt-Prigent, A., Agid, Y., Hirsch, E.C. Nitric Oxide Synthase and Neuronal Vulnerability in Parkinson's Disease. Neurosci 1996; 72 (2), 355-63.
Hu, S.H., Jiang, S-S., Yang, Y. Pioglitazone Ameliorates Intracerebral Insulin Resistance and Tau-Protein Hyperphosphorylation in Rats with Type 2 Diabetes. Exp Clin Endocrinol Diabetes 2013; 121: 220-4.
Johnson, G.V.W., Jope, R.S., Binder, L.I. Proteolysis of tau by calpain. Biochem and Biophys Res Commun 1989; 163(3), 1505–1511. Kaltschmidt, B., Linker, R.A., Deng, J., Kaltschmidt, C. Cyclooxygenase-2 is a neuronal target gene of NF-kappaB. BMC Mol. Biol 2002; 3(16).
Katayama, T., Imaizumi, K., Manabe, T., Hitomi, J., Kudo, T., Tohyama, M. Induction of neuronal death by ER stress in Alzheimer's disease. J Chem Neuroanatomy 2004; 28, 67-78.
Kelly, B.L., Ferreira, A., β-Amyloid-induced Dynamin 1 Degradation Is Mediated by Receptors in Hippocampal Neurons. J Biol Chem 2006; 281(38): 28079-28089.
Kerbler, G.M., Fripp, J., Rowe, C.C., Villemagne, V.L., Salvado, O., Rose, S., Coulson, E.J. Basal forebrain atrophy correlates with amyloid β burden in Alzheimer's disease. NeuroImage: Clin 2015; 7, 105-113.
Ko, H.S., von Coelln, R., Sriram, S.R., Kim, S.W., Chung, K.K.K., Pletnikova, O., et al. Accumulation of the Authentic Parkin Substrate Aminoacyl-tRNA Synthetase Cofactor, p38/JTV-1, Leads to Catecholaminergic Cell Death. J Neurosci 2005; 25 (35): 7968-78.
Lamarche, F., Carcenac, C., Gonthier, B., Cottet-Rousselle, C., Chauvin, C., Barret, L., et al. Mitochondrial Permeability Transition Pore Inhibitors Prevent Ethanol-Induced Neuronal Death in Mice. Chem Res Toxicol 2013; 26: 78-88.
Lei, P., Ayton, S., Finkelstein, D.I., Adlard, P.A., Masters, C.L., Bush, A.I. Tau protein: Relevance to Parkinson's disease. Int J Biochem Cell Biol 2010; 42: 1775-8.
Li, Y., Liu, L., Barger, S.W., Griffin, W.S.T. Interleukin-1 Mediates Pathological Effects of Microglia on Tau Phosphorylation and on Synaptophysin Synthesis in Cortical Neurons through a p38-MAPK Pathway. J. Neurosci. 2003; 23(5), 1605-11.
Liu, L., Chan, C. The role of inflammasome in Alzheimer's disease. Ageing Res Rev 2014; 15, 6-15.
Lucas, J.J., Hernandez, F., Gomez-Ramos, P., Moran, M.A., Hen, R., Avila, J. Decreased nuclear β-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3β conditional transgenic mice. EMBO J 2001; 20(1-2): 27-39.
Ma, Q-L., Yang, F., Rosario, E.R., Ubeda, O.J., Beech, W., Gant, D.J., Chen, P.P., Hudspeth, B., Chen, C., Zhao, Y., Vinters, HV., Frautschy, S.A., Cole, G.M. β-Amyloid Oligomers Induce Phosphorylation of Tau and Inactivation of Insulin Receptor Substrate via c-Jun N-Terminal Kinase Signaling: Suppression by Omega-3 Fatty Acids and Curcumin. Neurobiol of Disease 2009; 29(28), 9078-9089.
Mander, P., Brown, G.C. Activation of microglia NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: a dual-key mechanism of inflammatory neurodegeneration. J. Neuroinflammation 2005; 2(20).
Marchitti, S.A., Deitrich, R.A., Vasiliou, V. Neurotoxicity and Metabolism of the Catecholamine-Derived 3,4-Dihydroxyphenylacetaldehyde and 3,4-Dihydroxyphenylglycolaldehyde: The Role of Aldehyde Dehydrogenase. Pharmacol Rev 2007; 59 (2): 125-50.
Mehlen, P., Kretz-Remy, C., Preville, X., Arrigo, A.P. Human Hsp27, Drosophila Hsp27 and human alphaB-crystallin expression-mediated increase in glutathione is essential for the protective activity of these proteins against TNF-alpha-induced cell death. EMBO J. 1996; 15: 2695-706.
Mehlen, P., Schulz-Osthoff, K., Arrigo, A.P. Small stress proteins as novel regulators of apoptosis. Heat shock protein 27 blocks Fas/APO-1 and staurosporine-induced cell death. J Biol Chem 1996a; 271: 16510-4.
Miranda, S., Foncea, R., Guerrero, J., Leighton, F. Oxidative Stress and Upregulation of Mitochondrial Biogenesis Genes in Mitochondrial DNA-Depleted HeLa Cells. Biochem Biophys Res Commun 1999; 258 (1): 44-9.
Moon, Y.,Lee, K.H., Park, J-H., Geum, D., Kim, K. Mitochondrial membrane depolarization and selective death of dopaminergic neurons by rotenone: protective effect of coenzyme Q10. J Neurochem 2005; 93 (5): 1199-208.
Morris, J.K., Bomhoff, G.L., Gorres, B.K., Davis, V.A., Kim, J., Lee, P-P., et al. Insulin resistance impairs nigrostriatal dopamine function. Exp Neurol 2011; 231 (1): 171-80.
Morris, J.K., Zhang, H., Gupte, A.A., Bomhoff, G.L., Stanford, J.A., Geiger, P.C. Measures of striatal insulin resistance in a 6-hydroxydopamine model of Parkinson's disease. Brain Res 2008; 1240: 185-95.
Mota, S.I., Ferreira, I.L., Rego, A.C. Dysfunctional synapse in Alzheimer's disease – a focus on NMDA receptors. Neuropharm 2014; 76, 16-26.
Mudo, G., Makela, J., Di Liberto, V., Tselykh, T.V., Olivieri, M., Piepponen, P., et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson's disease. Cell Mol Life Sci 2012; 69 (7): 1153-65.
Nicholson, M., Ferreira, A. Increased membrane cholesterol might render mature hippocampal neurons more susceptible to β-Amyloid-induced calpain activation and tau toxicity. Journal of Neuroscience 2009; 29(14) 4640–4651. Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., Gaynor, K., Wang, L., LaFrancois, J., Feinstein, B., Burns, M., Krishnamurthy, P., Wen, Y., Bhat, R., Lewis, J., Dickson, D., Duff, K. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. PNAS 2005; 102(19), 6990-6995.
Oka, S., Kamata, H., Kamata, K., Yagisawa, H., Hirata, H. N-acetylcysteine suppresses TNF-induced NF-kappa B activation through inhibition of I kappa B kinases. FEBS Lett 2000; 472: 196-202.
Petit-Paitel, A., Brai, F., Cazareth, J., Chabry, J. Involvement of Cytosolic and Mitochondrial GSK-3β in Mitochondrial Dysfunction and Neuronal Cell Death of MPTP/MPP+-Treated Neurons. PLoS ONE 2009; 4 (5): 1-12.
Piao, Y., Kim, H.G., Oh, M.S., Pak, Y.K. Overexpression of TFAM, NRF-1, and myr-AKT protects the MPP+-induced mitochondrial dysfunctions in neuronal cells. Biochimica et Biophys Acta 2012; 1820 (5): 577-85.
Puchacz, E., Stumpf, W.E., Stachowiak, E.K., Stachowiak, M.K. Vitamin D increases expression of the tyrosine hydroxylase gene in adrenal medullary cells. Mol Brain Res 1996; 36(1): 193-6.
Ricote, M., Li, A.C., Willson, T.M., Kelly, C.J., Glass, C.K. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 1998; 391: 79-82.
Samuni, Y., Goldstein, S., Dean, O.M., Berk, M. The chemistry and biological activities of N-acetycysteine. Biochim et Biophys Acta 2013; 1830: 4117-29.
Sass, M.B., Lorenz, A.N., Green, R.L., Coleman, R.A. A pragmatic approach to biochemical systems theory applied to an α-synuclein-based model of Parkinson's disease. J Neurosci Methods 2009; 178(2): 366-77.
Shimura, H., Miura-Shimura, Y., Kosik, K.S. Binding of tau to heat shock protein 27 leads to decerased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem 2004; 279(17): 17957-62.
Snyder, C.M., Shroff, E.H., Liu, J., Chandel, N.S. Nitric Oxide Induces Cell Death by Regulating Anti-Apoptotic BCL-2 Family Members. PLoS ONE 2009; 4 (9).
Snyder, E.M., Nong, Y., Almeida, C.G., Paul, S., Moran, T., Choi, E.Y., Nairn, A.C., Salter, M.W., Lombroso, P.J., Gouras, G.K., Greengard, P. Regulation of NMDA receptor trafficking by amyloid-β. Nature Neurosci 2005; 8(8), 1051-1058.
Stutzmann, G.E., The pathogenesis of Alzheimer's disease—is it a lifelong "Calciumopathy"? Neuroscientist 2007; 13, 546-559.
Susin, S.A., Lorenzo, H.K., Zamzami, N., Marzo, I., Snow, B.E., Brothers, G.M., et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999; 397(6718): 441-6.
Takahashi, M., Yamada, T., Tooyama, I., Moroo, I., Kimura, H., Yamamoto, T., Okada, H. Insulin receptor mRNA in the substantia nigra in Parkinson's disease. Neurosci Lett 1996; 204 (3): 201-4.
Taylor, D.M., Moser, R., Regulier, E., Breuillaud, L., Dixon, M., Beesen, A.A., et al. MAP Kinase Phosphatase 1 (MKP-1/DUSP1) Is Neuroprotective in Huntington 's disease via Additive Effects of JNK and p38 Inhibition. J Neurosci 2013; 33(6): 2313-25.
Van Wagoner, N.J., Oh, J.W., Repovic, P., Benveniste, E.N. IL-6 production by astrocytes: autocrine regulation by IL-6 and the soluble IL-6 receptor. J. Neurosci 1999; 19, 5236-44.
Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev 2001; 15 (22): 2922-33.
Wang, Y., Shibasaki, F., and Mizuno, K. calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J. Biol. Chem.2005; 280,12683–12689 Winklhofer, K.F., Henn, I.H., Kay-Jackson, P.C., Heller, U., Tatzelt, J. Inactivation of Parkin by Oxidative stress and C-terminal Truncations: A Protective Role of Molecular Chaperones. J Biol Chem 2003; 278 (47): 199-208.
Xing, B., Liu, M., Bing, G. Neuroprotection with pioglitazone against LPS insult on dopaminergic neurons may be associated with its inhibition of NF-κB and JNK activation and suppression of COX-2 activity. J Neuroimmunol 2007; 192 (1-2): 89-98.
Xiong, N., Xiong, J., Khare, G., Chen, C., Huang, J., Zhao, Y., et al. Edaravone Guards Dopamine Neurons in a Rotenone Model for Parkinson's Disease. Plos One 2011; 6(6).
Yeager, M.P., Coleman, R.A. In silico evidence for glutathione- and iron-related pathogeneses in Parkinson's disease. J Neurosci Methods 2010; 188(1): 151-64.
Yu, S., Ueda, K., Chan, P. α-Synuclein and Dopamine Metabolism. Mol Neurobiol 2005; 31 (1-3): 243-54.
Zarubin, T., Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res 2005; 15 (1): 11-8.
Zheng, B., Zhixiang, L., Locascio, J.J., Lesniak, K.A., Roderick, S.S., Watt, M.L., et al. PGC-1α, A Potential Therapeutic Target for Early Intervention in Parkinson's Disease. Sci Transl Med 2010; 2 (52): 57-73.
Zhou, S., Yu, G., Chi, L., Zhu, J., Zhang, W., Zhang, Y., Zhang, L. Neuroprotective effects of edaravone on cognitive deficit, oxidative stress and tau phosphorylation induced by intracerebroventricular streptozotocin in rats. Neurotox 2013; 38: 136-45.
Zourlidou, A., Payne Smith, M.D., Latchman, D.S. HSP27 but not HSP70 has a potent protective effect against α-synuclein –induced cell death in mammalian neuronal cells. J Neurochem 2004; 88 (6): 1439-48.
Source: http://publish.wm.edu/cgi/viewcontent.cgi?article=1120&context=honorstheses
oral. Es hidrolizado rápidamente en el intestino a una tiolactona, la cual se convierte aquellos que presenten enfermedad renal terminal. La experiencia terapéutica en en el metabolito activo mediante un único paso de metabolismo por el citocromo pacientes con insuficiencia renal es limitada. P450, principalmente por las isoenzimas 3A4 y el 2B6 y en menor medida por las
PROGETTO UNIVA 2013 Journal Club Pietro Gareri, MD, PhD Geriatra ASP Catanzaro Lamezia Terme 3 Luglio 2013 Drug-induced parkinsonism (DIP) was recognized in the early 1950s as a commoncomplication of antipsychotic therapy; initially considered to be present in 4 - 40%of patients treated with the first neuroleptics